Синдром Прадера-Вилли, синдром Нунан (наследственные заболевания человека)


Общие сведения
Наследственные заболевания человека обусловлены различными изменениями генетической информации, образующимися на разных этапах фило/онтогенеза вследствие мутаций под воздействием эндо- и экзогенных причин. Мутации представляют собой нарушения количества/структуры наследственного материала и его функционирования на разных уровнях организации (хромосома, ген, геном) приводящими к появлению не присущих в норме новых признаков.
В основе наследственных болезней лежат герминативные мутации (возникшие в половых клетках). В отличие от них соматические мутации, происходящие в неполовых клетках человека, проявляются лишь у индивида, у которого они возникают и следующими поколениями не наследуются.
Процесс мутагенеза (формирования мутаций) происходит под действием мутагенов биологической, физической или химической природы:
- биологические мутагены представлены вирусами герпеса, кори, гриппа, краснухи; бактериальными токсинами, антигенами некоторых микроорганизмов;
- физические мутагены представлены ионизирующими излучениями (нейтроны, рентгеновское излучение, α-, β- и γ лучи), УФ-излучение; их особенностью является способность индуцировать мутации ионизирующим излучением в низких дозах, не вызывая при этом, лучевого поражения;
- химические мутагены — это большая группа химических соединений (тяжелые металлы, спирты, соли, кислоты и другие соединения); они широко распространены в среде обитания людей/сфере производства и содержатся в воздухе (сероводород, мышьяк, свинец, фтор, бензпирен и др.), почве (пестициды), воде, пищевых продуктах (табл. ниже).
Выраженной мутагенной активностью обладают многие лекарственные препараты (табл. ниже).
 Особенностью химических мутагенов является прямо пропорциональная зависимость их действия от дозы: чем доза мутагена выше, тем сильнее мутагенный эффект.
Особенностью химических мутагенов является прямо пропорциональная зависимость их действия от дозы: чем доза мутагена выше, тем сильнее мутагенный эффект.
 Различают:
Различают:
- индуцированные мутации, обусловленные воздействием вышеуказанных факторов, интенсивность воздействия которых превышает допустимые пределы;
- спонтанные мутации, которые проявиться без видимых внешних причин (спонтанно) под влиянием внутренних условий в организме в целом и в клетке в частности.
Список наследственных заболеваний человека чрезвычайно широкий. На сегодняшний день выявлено около 6 тыс. различных синдромов (совокупность внутренних/внешних функциональных и морфологических аномалий, вызванных одним фактором), обусловленных наследственным механизмом передачи. Некоторые генетические заболевания у детей и взрослых имеют одинаковую частоту встречаемости в различных странах мира, некоторые — определенную географическую/этническую и специфику распространения. Общая частота наследственных заболеваний в популяции варьирует в зависимости от нозологических единиц от 0,2 до 4%.
Зачастую наследственные заболевания смешивают с врожденными/семейными болезнями. Следует понимать, что «врожденные заболевания» и «наследственные болезни» не однозначные понятия. Врожденные — это любые заболевания наследственного/ненаследственного генеза, проявляющиеся сразу после рождения ребенка в то время, как наследственные болезни — это лишь те болезни, в основе которых структурные/количественные изменения в генетическом материале. Некоторые из них проявляются клинически сразу после рождения, некоторые — в юношеском, а иногда зрелом/пожилом возрасте.
Также существует различие между семейной и наследственной патологией, которое заключается в том, что семейная патология может быть, связана не с генетическими, а профессиональными/социально-бытовыми детерминантами. Существует ещё одно понятие в медицинской генетике — фенокопии, представляющее клинический синдром, который возникает в период эмбрионального развития под воздействием факторов внешней среды и по своим проявлениям сходный с наследственным заболеванием, но при этом он имеет негенетическую природу — внутриутробная гипоксия плода, инфекционные заболевания в период беременности (токсоплазмоз, краснуха), эндокринные нарушения, хронический алкоголизм родителей, тяжелая психическая травма и др.
Генетические заболевания у детей в большинстве случаев диагностируются сразу после рождения. Например, проявления внешних признаков синдрома Дауна настолько типичны, что диагноз ставится в родильном доме при первичном осмотре ребенка. Однако для установления точного диагноза требуется исследование кариотипа. Установлено, что около 25% наследственных болезней, обусловленных генными мутациями и практически все хромосомные болезни формируются в период внутриутробного развития и проявляются уже при рождении ребенка. Еще около 50% наследственных болезней, вызванных мутациями в генах, проявляется в первые три года жизни. В целом, почти 90% моногенно наследуемых болезней манифестируют к завершению периода полового созревания.
Некоторые хромосомные болезни, например, синдром Клайнфельтера, обусловленный увеличением у лиц мужского пола числа Х–хромосом обнаруживаются только в период полового созревания. Симптомы многих наследственных болезней обмена могут манифестировать в разном детском возрасте: фенилкетонурия — в первые месяцы жизни; целиакия (болезнь непереносимости злаков) — после введения в рацион питания каш во втором полугодии жизни; наследственные рахитоподобные заболевания на 2-3 годах жизни. Реже генные наследственные болезни проявляются в зрелом/пожилом возрасте (болезнь Паркинсона, Альцгеймера).
Такой разброс во времени обусловлен множеством факторов, в частности включению/выключению различных блоков генов в определенные периоды жизни (изменение гормонального фона при рождении ребенка, в периоды полового созревания). На проявление мутантного гена оказывают влияние (способствуют/препятствуют) и другие гены, что и определяет варьирование сроков манифестации наследственно обусловленной патологии в разные возрастные периоды.
Следует учитывать, что развитие многих наследственных синдромов/заболеваний не всегда обусловлено законами наследования. Степень вероятности их развития при определенной мутации характеризуется таким понятием как полная (ген в 100% реализуется в признак) и неполная (ген не всегда реализуется в признак) пенетрантность гена. То есть это понятие отражает вероятность развития наследственного синдрома, что позволяет оценивать риск как низкий, средний или высокий.
Естественный отбор как регулирующий фактор
Согласно данным литературы, частота генных мутаций варьирует в пределах 1-2/100 000 гамет. При этом, частота геномных/хромосомных мутаций значительно выше генных. В целом, общий уровень мутационного процесса является высоким, однако на ранних сроках развития отмечается высокая гибель гамет/зигот (около 50-70%), что, по-видимому, объясняется естественным отбором на всех стадиях внутриутробного развития генетически аномальных эмбрионов/плодов. Это проявляется на различных стадиях беременности внутриутробной гибелью зародышей/самопроизвольными выкидышами, преждевременными родами, а также мертворождениями и смертностью новорожденных после рождения в первые 7 суток. Так, «выбраковка» аномальных зародышей, обусловленных генетическими нарушениями, может достигать 40-50%, а максимальные репродуктивные потери характерны для первых 14 дней с момента зачатия (на стадии оплодотворения яйцеклетки/имплантации плодного яйца). Предположительно 50-60% зачатий из–за мутаций наследственных структур в беременность не реализуются, а заканчиваются гибелью зиготы до момента имплантации.
Поскольку наследственные болезни чрезвычайно разнообразны по типам мутаций, степени вовлеченности в процесс органов/систем, звеньям/видам нарушенного обмена, клинической симптоматике и характеру течения, что изложить их в пределах статьи практически невозможно. Поэтому рассмотрим лишь некоторые из них.
Патогенез
Патогенез наследственных болезней существенно различается в зависимости от локализации мутаций:
Патогенез генных болезней можно представить в виде основных звеньев: мутация гена → выработка патологически измененного белка (качественно/количественно) → нарушения биохимических процессов → изменения в клетках → изменения в органах → болезнь организма (рис. ниже).
 То есть, механизм развития генных болезней обусловлен изменениями на молекулярном, клеточном и органном уровнях, на каждом из которых он имеет свою специфику.
То есть, механизм развития генных болезней обусловлен изменениями на молекулярном, клеточном и органном уровнях, на каждом из которых он имеет свою специфику.
Патогенез хромосомных синдромов/болезней обусловлен нарушением структуры хромосом (хромосомные аберрации) или изменением числа хромосом (геномные мутации), реализуемые через различные механизмы, некоторые из которых представлены в таблице ниже.

Патогенез наследственных заболеваний нарушения обмена обусловлен конкретным механизмом расстройства метаболизма (углеводов, стероидов, аминокислот, липидов, пуринов и др.). Так развитие лизосомных болезней накопления вызвано дефектом единичных генов, контролирующих в макромолекулах процесс внутрилизосомного гидролиза. Такого рода мутации нарушают процесс синтеза/транспорта и созревания лизосомных ферментов, транспортных белков или белков-активаторов, подлежащих процессу гидролиза, что и приводит к накоплению недеградированных метаболитов.
Классификация
Единой классификации наследственных болезней до настоящего времени не существует. На практике используют три основных классификаций наследственных болезней: генная, патогенетическая и клиническая. В основу генетической классификации положен этиологический принцип (тип мутаций/характер их взаимодействия со средой) и соответственно выделяют несколько групп наследственных болезней.
Генные мутации
Обусловлены мутациями в одном гене. Молекулярной основой генных мутаций являются изменения состава/структуры нуклеотидов и регуляторных участков (повреждение структуры ДНК на уровне гена). Генные мутации разделяют на:
- мутации, вызванные заменой оснований в ДНК гена, на долю которых приходится около 20% спонтанных мутаций;
- мутации, которые обусловлены сдвигом рамки считывания, составляющих 80% всех спонтанных мутаций.
Мутации такого вида вызывают более выраженные нарушения в синтезируемых белках.
Мутагенный процесс может реализовываться различными механизмами: заменой нуклеотида, инсерцией/делецией нуклеотидов, внутригенной инверсией (рис. ниже).
 По типу наследования выделяют:
По типу наследования выделяют:
- аутосомно-доминантные мутации (синдром Марфана, болезнь Олбрайта, болезнь Реклингхаузена, несовершенный остеогенез, синдром Элерса-Данлоса и др.);
- аутосомно-рецессивные мутации (ихтиоз, фенилкетонурия, галактоземия);
- генные мутации, сцепленные с Х/Y-хромосомами (мышечная дистрофия Дюшенна, гемофилия, синдром Хантера, фосфат-диабет, болезнь Фабри).
В результате генной мутации на молекулярном уровне возможны различные варианты нарушений: синтез аномального белка; выработка генного продукта в избыточном/в уменьшенном количестве; отсутствие выработки первичного продукта.
Большинство болезней характеризуются клиническим полиморфизмом (многообразием фенотипических проявлений), что обусловлено различными факторами, оказывающими влияющими на пенетрантность (частоту проявления аллеля) и экспрессивность генов, вызывающих патологию. Примером генных болезней могут быть болезнь Гоше, муковисцидоз, гемофилия, синдром Марфана, алкаптонурия, миопатии Дюшенна-Беккера, фенилкетонурия, тирозинемия, мукополисахаридозы и др.
Хромосомные мутации
Возникают в результате хромосомных/геномных мутаций и представляют собой структурные изменения в отдельных хромосомах, в которые вовлекаются большое число генов, что вызывает изменения в нормальном диплоидном наборе. Выделяют внутри и межхромосомные мутации.
Внутрихромосомные мутации — это абберации (отклонения) в пределах одной хромосомы, представленные различными типами мутаций:
- Делеции — представляют собой утрату одного из внутреннего/терминального участков хромосомы, что может вызывать нарушение процесса эмбриогенеза и формирование различных аномалий развития (например, синдром «кошачьего крика»).

- Инверсии — образование фрагмента в результате разрывов хромосомы в двух точках, который после поворота на 180о встраивается на прежнее место, что приводит к нарушению последовательности расположения генов (перестройки генов).

- Дупликации — удвоение (умножение) участка хромосомы (трисомия, полисомия), что вызывает множественные пороки (задержку физического/психического развития, микроцефалию).

Межхромосомные мутации (транслокации) представляют собой обмен фрагментами между хромосомами:
- реципрокная транслокация – обмен своими фрагментами между двумя хромосомами;
- нереципрокная транслокация – транспортировка фрагмента одной хромосомы на другую;
- робертсоновская транслокация (центрическое слияние) – соединение двух хромосом в местах их центромеров, сопровождащееся потерей коротких плеч.
 То есть при делециях/дупликациях происходит изменение общего количества генетического материала, а степень фенотипического проявления мутаций непосредственно определяется размерами/значимостью изменяемых участков. В то время как при транслокациях/инверсиях не происходит изменение количества генетического материала, а только меняется его расположение.
То есть при делециях/дупликациях происходит изменение общего количества генетического материала, а степень фенотипического проявления мутаций непосредственно определяется размерами/значимостью изменяемых участков. В то время как при транслокациях/инверсиях не происходит изменение количества генетического материала, а только меняется его расположение.
Геномные мутации
Представляют собой изменения числа хромосом в геноме. К таким мутациям относятся:
- Анеуплоидия – представляющая собой изменение (увеличение/уменьшение) некратное гаплоидному числа хромосом в диплоидном наборе (2n-1/2n+1и т. д.). Подразделяется на моносомию (наличие лишь одной из двух хромосом). При моносомии невозможно нормальное развитие эмбриона и с жизнью несовместима.
- Трисомия (наличие в кариотипе трех гомологичных хромосом) — например, при болезни Дауна по 21-й паре, при синдроме Эдвардса по 18-й паре, синдроме Патау по 13-й паре.
- Полиплоидия – представляющая увеличение числа набора хромосом, которое кратное гаплоидному (3n, 4n, 5n и т.д.). Относится к летальным мутациям.
 Наиболее часто встречаются такие хромосомные болезни, как синдромы Дауна, Шерешевского-Тернера, Патау, Эдвардса, синдром «кошачьего крика», Клайнфельтера и др. Однако, хромосомные болезни не исчерпывают весь список последствий различного рода перестроек в хромосомном аппарате. Установлена связь между хромосомными аномалиями и возникновением злокачественных образований.
Наиболее часто встречаются такие хромосомные болезни, как синдромы Дауна, Шерешевского-Тернера, Патау, Эдвардса, синдром «кошачьего крика», Клайнфельтера и др. Однако, хромосомные болезни не исчерпывают весь список последствий различного рода перестроек в хромосомном аппарате. Установлена связь между хромосомными аномалиями и возникновением злокачественных образований.
Так, генные хромосомные перестройки RET/PTC связаны с раком щитовидной железы. Ген RET кодирует рецепторную тирозинкиназу — это метаболический рецептор, представляющий собой белок и имеющий тирозинкиназную активность. Рецепторные тирозинкиназы являются основными регуляторами процессов в клетке и имеют значение в развитии злокачественных опухолей. Мутации рецепторных тирозинкиназ активируют ряд сигнальных каскадов, вызывающих изменения белков. Распространенные типы перестройки — хромосомные аберрации RET/PTC1 и RET/PTC3. Установлено, что 20% случаев папиллярного рака щитовидной железы связано с транслокациями RET/PTC. Эти перестройки выявляются у детей и лиц молодого возраста, которые перенесли облучение. В 50% случаев мутация RET связана с семейной формой медуллярного рака щитовидной железы.
Наиболее значимые хромосомные аномалии, которые оказывают влияние на возникновение/рост новообразований: транслокации, делеции, рецессивные мутации, амплификации.
Мультифакториальные болезни (болезни с наследственной предрасположенностью)
Заболевания этой группы обусловлены комбинированным действием генетических факторов риска, которые формируют наследственную предрасположенность к заболеванию и неблагоприятных внешних факторов. Они представлены большой группой заболеваний преимущественно с хроническим течением, включая нервно-психические, сердечно-сосудистые, онкологические, эндокринные, иммунные (атеросклероз, врожденные пороки развития, гипертоническая болезнь, ИБС, язвенная болезнь желудка, некоторые формы сахарного диабета, бронхиальная астма, шизофрения и множество других).
Базисом наследственной предрасположенности к болезням является генетический полиморфизм (генетическое разнообразие) человеческой популяций по структурны/транспортным белкам, ферментам, антигенным системам. Заболевания этой группы вносят чрезвычайно большой вклад в заболеваемость и смертность населения, и часто являются причиной инвалидизации.
Патогенетическая классификация
Кроме генетической классификации частот используется патогенетический принцип классификации наследственных болезней, согласно которой выделяют группу наследственных болезней накопления в основе которой лежит недостаточность ферментов, принимающих участие в метаболизме веществ, недостаточность транспортных белков, белков иммунной системы, каналов и рецепторов. Согласно этой классификации, выделяют несколько групп заболеваний.
Наследственные нарушения обмена:
- обмена углеводов (например, галактоземия, гликогеновая болезнь и др.);
- обмена стероидов (например, дреногенитальный синдром и др.);
- обмена аминокислот (фенилкетонурия и др.);
- обмена липидов (например, болезнь Гоше, Ниманна-Пика и др.);
- обмена пуринов/пиримидинов (например, синдром Леша-Найяна и др.);
- обмена соединительной ткани (например, синдром Марфана/мукополисахаридозы и др.);
- обмена в эритроцитах (например, анемия Минковского-Шоффара и др.);
- всасывания в пищеварительном тракте (например, непереносимость лактозы, муковисцидоз, целиакия и др.);
- лизосомные болезни накопления.
Врожденные пороки развития. Включают полное/частичное отсутствие органа/его части (аплазия), недоразвитие органа (гипоплазия), уменьшение его размеров (гипоплазия), избыточное развитие органа (макросомия), необычное расположение органа (эктопия), закрытие/заращение естественных каналов/отверстий (атрезия), увеличение количества органов, сращение отд. органов между собой и др.
Комбинированные состояния — характеризуются сочетанными нарушениями метаболизма.
В качестве примера из этой группы рассмотрим лишь митохондриальные генные болезни и лизосомные болезни накопления.
Митохондриальные генные болезни
Митохондриальные нарушения представляют собой наследственные синдромы, обусловленные мутациями в мтДНК (митохондриальной ДНК) или генах, расположенных на хромосомах ядре клетки генов, которые являются ответственными за митохондриальные белки (синдромы Кернса-Сейра, NARP, Барта, Пирсона, синдром Ли, болезнь Лебера и др.). Иными словами — это обширная группа патологических состояний, которые обусловлены генетическими, биохимическими и структурными дефектами митохондрий, проявляющиеся нарушением тканевого дыхания и недостаточностью энергетического обмена.
Кроме первичных заболеваний, характеризующихся митохондриальной недостаточностью, существует множество заболеваний, включающих в качестве «вторичных» патогенетических звеньев нарушения клеточного энергообмена: заболевания соединительной ткани, гипопаратиреоз, синдром хронической усталости, кардиомиопатии, мигрени, гликогенозы, рахит, диабет, тубулопатии, печеночная недостаточность и др.
Наиболее остро мутации мт-ДНК сказываются на энергозависимых (мышечной/нервной) тканях и очень часто проявляются в форме различных миопатий/невропатий/кардиопатий (нейропатия Лебера, синдром Пирсона, melas-синдром и др.), сопровождающие умственной отсталостью, энцефалопатией, ранней смертностью. Ниже на рисунке приведен перечень органов-мишеней, затрагиваемых митохондриальными нарушениями.
 Передача митохондриальных заболеваний происходит только по материнской линии, что обусловлено передачей в зиготу своих митохондрий только яйцеклеткой.
Передача митохондриальных заболеваний происходит только по материнской линии, что обусловлено передачей в зиготу своих митохондрий только яйцеклеткой.
Лизосомные болезни накопления
Большинство из них развиваются вследствие дефекта определенных генов, кодирующих ферменты, которые участвуют в процессе превращения субстратов в продукты. В основе таких расстройств лежит процесс накопления продуктов, обладающих токсическим действием или способных нарушать процессы метаболизма (синтеза жизненно важных соединений). Лизосомные болезни накопления — это редкие заболевания, суммарная частота которых составляет 1:8000 новорожденных. Примером таких заболеваний являются мукополисахаридозы, сфинголипидозы, муколипидозы, гликопротеинозы, болезнь Гоше, гранулематоз Фарбера, болезнь Ниманна-Пика и др.
Причины
Общим этиологическим фактором развития наследственных болезней являются мутации на различных уровнях в генах, хромосомах и геноме, которые затрагивают структурные, транспортные и эмбриональные белки, а также ферменты. При этом каждое наследственное заболевание обусловлено конкретными генетическими нарушениями. Например, перечисленные ниже.
Синдром Прадера-Вилли — обусловлен повреждением хромосомы 15 (локуса q11.2-q13). Хромосомная аномалия реализуется в момент обмена генетических материалов отца/матери при оплодотворении яйцеклетки. В 70% случаев мутация вызвана дефектом отцовской 15 хромосомы, со стороны отца и реже (в 30%) наследованием обеих 15 хромосом по материнской линии.
Синдром Ваарденбурга — в зависимости от варианта патологии мутации происходят в различных генах: при синдроме Ваарденбурга 1-го типа — мутации на 2-й хромосоме в гене PAX3; синдром Ваарденбурга 2-го типа подкласса «a» обусловлен дефектами гена MITF на 3-й хромосоме, Синдром Ваарденбурга подкласса «d» вызван мутациями на 8-й хромосоме в гене SNAI2; синдром Ваарденбурга 3-го является наиболее тяжелым вариантом и его причиной являются мутации гена PAX3, при этом дефектные формы гена находятся в гомозиготном состоянии; синдром Ваарденбурга 4-го типа подкласса (a, b, c), вызывается мутациями различных генов: подтип «а» обусловлен мутацией на 13-й хромосоме в гене EDNRB, который кодирует последовательность белка В-эндотелина; подтип «b» синдрома Ваарденбурга вызывается дефектами гена EDN3, который локализуется на 20-й хромосоме, а подтип «c» вызван мутацией гена SOX10, расположенный на 22-й хромосоме.
Синдром Блума возникает в результате мутаций гена BLM (15q26.1), кодирующего ДНК-хеликазу, фермент, осуществляющий поддержку геномной целостности (стабильности ДНК в процессе репликации). Мутации гена BLM дезактивируют белок, который кодируется геном BLM и нарушают процесс экспрессии.
Синдром Нунан — известны 9 генов (PTPN11; SOS1; RAF1; KRAS; NRAS; BRAF; SHOC2; CBL; RIT1) мутация в которых приводит к развитию синдрома Нунан: эти гены кодируют белки/компоненты Ras/MAPK-пути и приводят к его активации.
Дефицит альфа1-антитрипсина обусловлен недостаточностью А1АТ в сыворотке крови. Гликопротеин А1АТ кодируется геном SERPINA1, который локализуется в хромосоме 14q32.1 на ее длинном плече в локусе ингибитора протеиназы.
Причинами болезней накопления также являются дефекты в различных структурах генома. Так муколипидозы II и III вызваны мутациями генов GNPTAB/GNPTG, которые фермент N-ацетилглюкозамин/фосфотрансферазу, что приводит недостаточности энзима и вызывает нарушение процесса присоединения к лизосомным ферментам маннозо-6-фосфата. В основе развития муколипидоза IV типа — нарушением работы кальциевого канала, обусловленного мутаций на коротком плече хромосомы 19 в гене MCOLN1 (19p13.2).
Некоторые хромосомные аномалии вызывают множественную патологию. Так, делеция в длинном плече 22 хромосомы (del 22q11DS) манифестирует различными клиническими проявлениями: аномалиями развития крупных сосудов, врожденными пороками сердца, врожденными лицевыми аномалиями, иммунным/эндокринными нарушениями, а также может проявляться различным функциональным/психическими ассоциациями, приводящее к нарушению психики (синдром гиперактивности/дефицита внимания, генерализованное тревожное расстройство, депрессии, расстройства аутистического спектра, фобии, шизофрения), а также к задержке речевого и психомоторного развития детей.
Симптомы наследственных заболеваний человека
Семиотика наследственной патологии характеризуется большим разнообразием черт дизэмбриогенеза, патологических симптомов и малых аномалий развития, нарушающих функции/структуру как отдельных органов, систем, так и всего организма. Рассмотрим симптоматику лишь некоторых из них.
Синдром Ваарденбурга
В клинике чаще всего проявляется телекантом (смещение внутреннего угла глаза), частичным альбинизмом (пигментные аномалии — обесцвечивание кожи, волос, глаз) и врожденной нейросенсорной тугоухостью. У больных редко выявляется все характерные признаки, кроме того, каждый симптом имеет разную степень выраженности. При данном синдроме часто наблюдаются: сросшиеся брови, широкая переносица, гетерохромия радужки (разный цвет глаз или имеется сектор другого цвета в одной и той же радужной оболочке). Эти проявления могут сочетаться с аномалиями рук или с болезнью Гиршпрунга (аномалия развития толстой кишки).
В некоторых случаях присутствуют опущение века, расщелина неба, выступающая челюсть, пороки сердца. Патология рук включает слияние костей запястья, недоразвитие кистей и мышц, ограничение подвижности. Со стороны позвоночника отмечаются сколиоз, сильный поясничный лордоз и добавочные позвонки. В зависимости от дополнительных признаков в клинике выделяют 4 типа заболевания.
Первый тип считается классическим, сочетает телекант, пигментные нарушения, тугоухость и аномалии лица. Третий тип протекает более манифестно и тяжело, поскольку тугоухость, нарушения пигментации и пороки лица сильно выражены. В дополнение к этому у больных развивается гипоплазия мышц (больше верхних конечностей).
Синдрома Ваарденбурга 4-го типа дополнительно включает проявления болезни Гиршпрунга. У больных появляются запоры, вздутие, боли в животе и расширение ободочной кишки. Выраженность этих симптомов зависит от длины кишки, в которой отсутствует нервное сплетение. У некоторых больных при этом типе может быть, нарушена миелинизация нервных волокон, что сопровождается болями, изменением чувствительности и парезами.
Синдром Прадера-Вилли
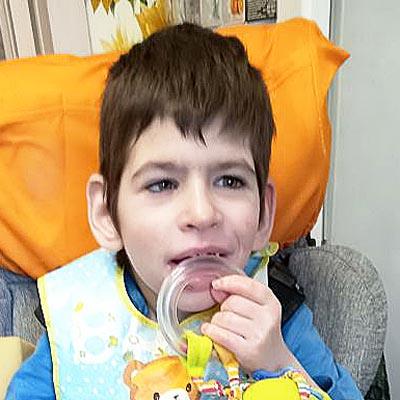
Дети с данной патологией сразу после рождения отличаются от здоровых — они рождаются доношенными, но с явлениями внутриутробной гипотрофии (пониженного питания) и асфиксии, они вялые и неактивные, не кричат. Это генетическое заболевание человека, передающееся по наследству, протекают в две фазы: первая отмечается у детей до 1-1,5 лет и протекает со значительной мышечной гипотонией, снижением сосательного рефлекса и глотательного рефлекса, поэтому кормление ребенка затруднено. Первые месяцы такие дети почти неподвижны из-за сниженного тонуса мышц и у них отмечается повышенная сонливость. Если они не могут самостоятельно дышать, то жизнедеятельность их поддерживается аппаратами. Симптомы синдрома Прадера-Вилли второй фазы проявляются к двум-трем годам и характеризуются чрезмерным чувством голода и повышенным аппетитом. У больных отсутствует чувство сытости, что приводит к ожирению (отложение жира происходит на туловище и конечностях).
Родители узнают о диагнозе поздно — когда развивается уже выраженное ожирение, грозящее задержкой и остановкой дыхания во время сна. Мышечная гипотония уменьшается к школьному возрасту и может исчезнуть. У больных отмечаются диспропорционально маленькие стопы и кисти, а также другие микроаномалии: высокий лоб, миндалевидные глаза, тонкие, опущенные веки, маленькая узкая голова, выворот века, недоразвитие челюсти. Кожа и волосы у больных детей светлее, чем у родственников и отмечается также гипопигментация радужки. Рост меньше положенного по возрасту. Характерным проявлением синдрома является гипогонадизм — недоразвитие полового члена и мошонки, недоразвитие половых губ и матки.
Встречается также у больных сахарный диабет. По психомоторному развитию ребенок отстает от нормы, речь его затруднена, а словарный запас небольшой. У детей ухудшены зрительная и слуховая память, а также концентрация внимания. Настроение у таких больных часто меняется. При синдроме Прадера-Вилли отмечается высокий риск развития лейкемии. Есть форум, посвященный данной теме, а силами родителей больных детей учрежден первый в России благотворительный фонд, который помогает детям с этим заболеванием. Существует общий чат родителей, где они получают информацию, моральную поддержку и делятся своим опытом ухода за больными детьми. Фондом оказывается адресная помощь.
Синдром Блума
Синонимом является врожденная телеангиэктатическая эритема. Это связано с тем, что в первые недели после рождения на щеках, носу и тыле кистей появляются пузыри и эритема (покраснение). Телеангиэктатическая эритема может появиться в течение первого года жизни и провоцирует ее воздействие ультрафиолета. На лице она имеет вид бабочки.
Учитывая то, что больные отличаются повышенной чувствительностью к ультрафиолету, кожные проявления под его действием усиливаются и прогрессируют. непродолжительное нахождение на солнце приводит к повреждениям кожи различной тяжести. После восстановления кожи образуются гиперпигментированные или светлые пятна, а также участки атрофии.
При рождении дети имеют небольшой вес (1900-2000 г) и потом плохо его прибавляют и медленно растут. Больные во взрослом состоянии невысокого роста (не более 150 см). Психическое развитие соответствует возрасту.
Половое созревание у них задерживается. У мужчин отмечается бесплодие и ранняя менопауза у женщин, кроме того, многие из них не могут забеременеть.
Другие характерные проявления данной патологии:
- В младенчестве очень тонкая жировая клетчатка, поэтому дети выглядят худощавыми.
- Кровотечение из губ.
- Специфические черты лица: узкое и длинное лицо, микрогнатия (маленькая челюсть), большой выступающий нос и уши.
- Повышенная восприимчивость к инфекциям. В детстве часто возникают инфекции дыхательных путей (рецидивирующие бронхиты, пневмонии, отиты). Частые инфекции дыхательных путей постепенно приводят к развитию обструктивной болезни.
- Аномалии и заболевания глаз (гипоплазия зрительного нерва и конъюнктивит).
- Снижение концентрации внимания и памяти.
Есть еще одна особенность лиц, страдающих этим синдромом — повышенная предрасположенность к онкологическим заболеваниям. Причем они развиваются в молодом возрасте и может обнаруживаться несколько первичных опухолей у одного человека. Лейкемии и лимфомы характерны для детского возраста, а карциномы толстой кишки, пищевода, груди встречаются у взрослых. В местах, доступных солнцу, часто развиваются плоскоклеточный рак кожи и базалиома. У 15% пациентов развивается сахарный диабет.
Синдром Нунан
Данный синдром отличается множественными врожденными аномалиями. Клиническая картина напоминает синдром Шерешевского-Тернера. Симптомы включают черепно-лицевые аномалии, задержку роста и интеллектуального развития разной выраженности. Характерным является врожденная патология сердечно-сосудистой системы. Наиболее часто (до 90% случаев) встречается стеноз клапана легочной артерии, дефект межпредсердной перегородки и гипертрофическая кардиомиопатия. Также у больных описаны дефекты митрального клапана, межжелудочковой перегородки, аортальный стеноз и пороки магистральных сосудов. Дети имеют перепончатую шею, широко расставленные глаза и опущенные веки. У них отмечаются низко посаженные уши, выпуклое нёбо и укороченный 4-й палец кисти.
У детей может быть нарушение слуха. У мальчиков отмечаются недоразвитые яички или крипторхизм (не опустившиеся яички). У мальчиков и девочек задерживается половое созревание, юноши часто имеют низкий уровень тестостерона и бесплодны. Менструация у девочек начинается позже, но фертильность не нарушается, и они могут иметь детей.
Из других симптомов встречаются:
- Отставание в росте.
- Низкий рост.
- Плоская грудная клетка.
- Задержка психического развития.
- Почечные аномалии.
- Патология глаз.
Дефицит альфа-1-антитрипсина
Альфа1-антитрипсин — ингибитор протеолитических ферментов (в частности, эластазы), который защищает легочную ткань от влияния ферментов и деструкции (разрушения). При дефиците альфа1-антитрипсина происходит повышение активности протеолитических ферментов и в результате этого в эластичных волокнах дыхательных путей происходят деструктивные процессы. Легочная ткань теряет эластичность и развиваются патологические состояния — обструктивные бронхиты, эмфизема, бронхоэктазы, идиопатический фиброз, повторные пневмонии, рак легкого. При низком уровне этого белка клинические признаки заболевания могут полностью отсутствовать или проявляются незначительно.
У взрослых преобладают формы с поражением легких. Клинические проявления поражения дыхательной системы начинаются в возрасте 30–40 лет. У больных возникает одышка при физической нагрузке, которая быстро прогрессирует, периодически появляется свистящее дыхание и кашель с мокротой. Постоянный кашель встречается при бронхоэктазах. У курильщиков легкие вовлекаются в процесс раньше, нежели у некурящих и заболевание быстрее прогрессирует.
Также имеется связь дефицита А1АТ с заболеваниями печени. Поражение печени чаще отмечается у детей и проявляется желтухой, увеличением печени и повышением ферментов печени уже в первую неделю жизни. В 2-4 месяца желтуха исчезает. В 20% случаев такие поражения печени приводят к циррозу печени уже в детские годы или развивается цирроз во взрослом возрасте. Также увеличивается риск гепатоцеллюлярного рака печени. Не исключается и поражение легочной ткани, которые проявляются в 2-3 года периодической бронхиальной обструкцией, а 7-летнему возрасту уже развивается эмфизема легких.
Другие нарушения, связанные дефицитом альфа1-антитрипсина — это кровотечения, синяки, неспецифический язвенный колит, панникулит (воспаление подкожно жировой клетчатки), аневризмы (расширение артерий) различных локализаций и гломерулонефриты.
По клиническому течению выделяют следующие формы:
- дефицит А1АТ с поражением дыхательной системы;
- с поражением печени;
- с поражением печени и легких.
Муколипидоз
Болезни накопления — это редкие генетические заболевания, которые не до конца изучены. Муколипидоз относится к таким заболеваниям, характеризуется прогрессирующим течением и с возрастом клинические проявления становится более выраженными. Известно несколько типов — муколипидоз II типа и муколипидоз III типа с подтипами IIIА и IIIС.
Муколипидоз II типа проявляется уже с рождения отличаются выраженными аномалиями скелета: контрактуры суставов, деформация стоп, короткая грудная клетка и шея, врожденный вывих бедра, множественные переломы костей. Характерны также аномалии лица: мелкие глазницы, выпученные глаза, отекшие веки, грубые черты лица и гиперплазия десен.
Новорожденные плохо набирают вес и растут. В дальнейшем дети очень отстают в росте, а также у них отмечается тяжелая умственная отсталость. У большинства из них регистрируется поражение сердца. Первые признаки поражения сердца проявляются у новорожденных кардиомегалией и сердечной недостаточностью. У детей старшего возраста развивается поражение митрального и аортального клапана. Прогрессирующее течение приводит к смерти детей в первые десять лет жизни.
Муколипидоз IIIA типа отличается от муколипидоза II типа менее тяжелым течением и проявляется в возрасте двух лет. Дети имеют низкий рост, укороченное туловище и руки, помутнение роговицы, грубые черты лица. Часто отмечается тугоподвижность суставов, искривление позвоночника и утолщение ключиц. Так как и при II типе отмечается патология сердечно-сосудистой системы — чаще всего повреждение аортальных клапанов (аортальная недостаточность или аортальный стеноз). Интеллект у детей нормальный, а у 50% незначительно снижен. Этот тип муколипидоза имеет благоприятный жизненный прогноз — больные доживают до пожилого возраста. Самая легкая и доброкачественная форма — муколипидоз IIIC типа.
Генетическое заболевание Эдди
Второе название этой патологии — эктодермальная дисплазия. Является малоизученным генетическим заболеванием. Понятие дисплазия включает разные пороки органов и тканей, возникающие при внутриутробной закладке. Аномалии касаются закладки зубов, эпидермиса, потовых желез, волос, ногтей. Эктодермальной дисплазией болеют только мальчики, а девочки являются носителями гена, но у них могут быть минимальные проявления.
В разных сочетаниях у больных при синдроме Эдди выявляются:
- Редкие и тонкие волосы, которые медленно растут, ресницы и брови короткие и редкие. Возможно полное выпадение волос и отсутствие бровей и ресниц.
- Тонкая мелкоморщинистая кожа, кожа в области вокруг глаз и рта особенно истончена, может быть пигментирована.
- Позднее прорезывание зубов (в возрасте 2-3 лет), сроки прорезывания зубов нарушены. У детей долго сохраняются молочные зубы. Постоянные зубы деформированы, большие промежутки между зубами. Зубы или не в полном количестве или полностью отсутствуют. Отмечается аномальная шиповидная форма передних зубов с клыкообразным окончанием.
- Гипоплазия потовых желез, поэтому отсутствует потоотделение, что влечет нарушение терморегуляции — больные склонны к перегреванию и повышению температуры.
- Дисплазия лицевого черепа, что придает больным черты старческий вид: большой лоб и мозговой череп, выступающие лобные бугры и надбровные дуги, седловидный нос, маленькие крылья носа, запавшая переносица, часто запавшие щеки и деформированные уши. Старческий вид больных связан с полным отсутствием зубов и уменьшением высоты нижней челюсти. За счет деформации или отсутствия зубов вокруг рта образуются складки, что усугубляет старческий вид. Особенности лица у разных больных могут быть выражены в разной степени или могут почти отсутствовать.
- Гипоплазия слизистых желез во рту сопровождается уменьшением слюноотделения, поэтому у больных имеется предрасположенность к стоматитам и хейлитам (трещины на кайме губ). Из-за уменьшенной выработки слизи в глотке и гортани у больных появляется грубый и хриплый голос. Снижение выработки трахеобронхиального секрета вызывает частые трахеобронхиты.
- Расщелины губы и неба.
- Гипоплазия слезных желез, поэтому отмечается недостаточная выработка слезной жидкости (плачут без слез) и часто развиваются кератиты, блефариты, конъюнктивиты, светобоязнь.
- Аномалии кистей и стоп: эктродактилии (недоразвитие пальцев) и синдактилии (неполное или полное сращение пальцев).
- Экзема.
- Иммунодефицит.
Анализы и диагностика
Современные методы диагностики генетических заболеваний базируются на использовании различных молекулярно-биологических технологий и ДНК-маркеров, что позволяет выявить субмикроскопическую/функциональную патологию на уровне ДНК у больных даже без видимой хромосомной патологии:
- Молекулярно-генетические методы диагностики (FISH, ПЦР, SKY) позволяют обнаружить наследственные синдромы еще до рождения ребенка.
- Биохимические методы эффективны при наследственных болезнях обмена (гликогеновая болезнь, фенилкетонурия, болезни пуринового обмена и др.), позволяющие выявить продукты несовершенного метаболизма.
- Цитогенетический метод (изучение состояние хромосом) проводится для ранней диагностики патологий детей/выявления причин нарушения репродуктивной функции.
- Метод выявления гетерозиготного носительства — используется для установления носительства родителями и определения вероятности их проявления у ребенка.
- В диагностике наследственных болезней значительное место занимает неонатальный скрининг новорожденных, который проводится в первые 10 дней жизни ребенка. При обязательном скрининге перечень патологий сокращен до пяти: гипотериоз, муковисцидоз, андрогенитальный синдром, фенилкетонурия, галактоземия.
Лечение
Можно выделить три подхода к лечению болезней с наследственной предрасположенностью: этиологическое, патогенетическое и симптоматическое. Значительно реже используется хирургические методы лечения, например, реконструктивная хирургия при расщелине неба, губы, стенозе привратника, накладывание анастомоза между нижней полой и воротной венами и т.д.
Этиотропное лечение направлено, если это возможно, на устранение причины. Генетические дефекты корректируются терапией — внесением в клеточный геном органов, которые поражены, нормально экспрессируемого «здорового гена» и он выполняет функцию аномального гена.
Патогенетическая терапия разрывает звенья патогенеза. Это может быть:
- Заместительная терапия — введение препаратов дефицитного вещества, который не синтезируется в организме из-за той или иной аномалии гена. Большинство лизосомных болезней накопления — это ферментопатии. На сегодняшний день генной инженерией созданы ферменто-заместительные препараты лишь при некоторых лизосомных болезнях.
- Коррекция метаболизма — с питанием ограничиваются те вещества, которые не усваиваются в организме (фенилаланин, лактоза), выводятся метаболиты, накапливающиеся в избытке, а также регулируется активность ферментов.
- Хирургическая коррекция дефектов (создание шунта у больных при гепатотропных гликогенозах).
Симптоматическая терапия устраняет отдельные симптомы, которые усугубляют состояние больного. Сюда относится массаж, ортопедическая коррекция, хирургическое удаление перемычек между пальцами, удаление лишних пальцев, ортодонтическая (стоматологическая) коррекция, пластические операции и вмешательства при пороках сердца и сосудов, а также назначение препаратов гормонов роста при отставании в росте и половых гормонов при гипогонадизме. При некоторых наследственных синдромах возможно этиотропное и патогенетическое лечение, а при других проводится только симптоматическое.
Эффективное лечение синдрома Прадера-Вилли — введение рекомбинантного гормона роста. При нарушении биоэнергетического обмена назначаются курсами метаболические препараты: Кудесан (коэнзим Q10), Когитум (общетонизирующий препарат на основе янтарной кислоты), Янтавит (янтарная кислота).
Больным показан массаж и при необходимости ортопедическая коррекция. В подростковом возрасте назначают гонадотропины (фолликулостимулирующий и лютеинизирующий гормон, препараты андрогенов у юношей и эстроген-прогестагенных у девушек), которые способствуют развитию вторичных половых признаков. Важно строго контролировать питание ребёнка и повысить его физическую активность. Также необходимо проводить занятия с логопедом, дефектологом, лечиться у эндокринолога.
Лечение синдрома Ваарденбурга направлено на устранение симптомов. Консервативное лечение глухоты неэффективно, поэтому очень рано прибегают к хирургическому лечению — имплантируют кохлеарный имплант с целью стимулирования слухового нерва. В редких случаях выполняется косметическая операция по поводу телеканта. Так как при пигментных аномалиях кожи больные чувствительны к солнцу и у них часто бывают солнечные ожоги, а также развивается рак кожи, важно избегать солнечного света, необходимо пользоваться солнцезащитными средствами. При глазных аномалиях рекомендуется ношение очков с тонированными стеклами. Гипоплазия мышц несколько уменьшаться при занятиях гимнастикой и применении физиотерапевтического лечения. При аномалиях верхних конечностей (слияние костей запястья и недоразвитие кистей) проводится ортопедическое лечение.
При синдроме Блума также проводится симптоматическое лечение. Из-за повышенной чувствительности к ультрафиолету и риском возникновения опухолей кожи не рекомендуется пребывание на солнце, а также важно использовать солнцезащитный крем. Больным нужно избегать рентгенологических исследований. Для лечения частых бактериальных инфекций применяются антибиотики. Важно своевременное наблюдение и обследование по поводу онкологических заболеваний.
При дефиците альфа1-антитрипсина применяется специфическое замещающее лечение препаратами альфа-1-антитрипсина, которые получают из донорской плазмы. Препараты Земайра, Араласт, Глассиа применяются внутривенно. Они оказывает противовоспалительное действие и замедляют прогрессирование эмфиземы. Заместительную терапию не рекомендуется проводить больным, которые продолжают курить или имеют поражение печени, связанное с дефицитом А1АТ.
Лечение синдрома Эдди направлено на устранение неблагоприятных симптомов. Для предотвращения пересыхания полости носа, рта и глаз рекомендуется частое орошение слизистых и использование «искусственной слезы», что предотвращает повреждение роговицы. Детей нужно оберегать от воздействия солнца и высокой температуры, не допуская перегревания. При отсутствии зубов рекомендуется постоянное ношение съемных протезов, что нормализует функцию жевания, восстановит форму нижней трети лица и улучшит внешний вид.
Синдром Нунан неизлечим. Хирургическое лечение заключается в устранении пороков сердца. Проводится консервативное лечение, которое включает:
- для стимуляции роста введение препаратов гормона роста;
- ноотропные средства;
- психотропные препараты;
- при гипогонадизме — препарат Хориомон (хорионический гонадотропин) и Тестостерон.
Доктора



Лекарства
 Растан
Растан Джинтропин
Джинтропин- Препараты альфа-1-антитрипсина: Земайра, Араласт, Глассиа.
- Препараты гормона роста: Генотропин, Растан, Джинтропин, Соматоропин.
Процедуры и операции
При эмфиземе больным с дефицитом А1АТ проводится операция по уменьшению объема легких, а также устанавливаются эндобронхиальные спирали или эндобронхиальные клапаны. Их помещают в дыхательные пути с помощью бронхоскопа. При эмфиземе проводится трансплантация легких. Больные с терминальной стадией цирроза печени также рассматриваются как кандидаты для трансплантации печени. Донорская печень после пересадки синтезирует нормальное количество альфа1-антитрипсина.
Диета
Какой-либо единой диеты при наследственных болезнях нет, что обусловлено их разнообразием. Вместе с тем диетическое питание является составляющим элементом при некоторых заболеваниях, в частности, болезней обмена, при которых отсутствуют конкретные ферменты.
Особенности питания важны для детей с синдромом Прадера-Вилли. Важен контроль размера порций и ограничение доступности пищи, поскольку дети могут прятать ее, выпрашивать у незнакомых людей и т.д. В дошкольном возрасте не рекомендуются постоянные строгие ограничения в питании, чтобы обеспечить процессы роста и развития; в младшем школьном возрасте показано назначение гипокалорийной сбалансированной диеты (1000–1200 калорий в сутки) с добавлением витаминов и кальция – под контролем диетолога; ограничение доступности пищи дома и в школе (в том числе использование «запирающихся» шкафов и холодильников, маленьких тарелок). Поощрение физической активности дома и в школе играет важную роль в коррекции массы тела. В настоящее время не существует эффективных лекарственных препаратов для коррекции гиперфагии, поэтому уменьшение поступления калорий и доступности пищи — единственная доступная стратегия для предотвращения или ограничения набора массы тела при СПВ.
При фенилкетонурии показана специальная диета, исключающая продукты, содержащие фенилаланин — арахис, арахисовая паста, фасоль, бобы, соя, яичный порошок, сыр Пармезан, брынза и другие сыры, горбуша, треска, говядина, индейка, икра красная. Дети с фенилкетонурией рождаются без проявлений болезни, но из-за поступления фенилаланина с молоком проявляются клинические проявления. Если кормящая женщина придерживается диеты и в дальнейшем из питания ребенка исключаются эти продукты, то это предупреждает развитие клинической картины.
Больным с поражением печени на фоне дефицита А1АТ, необходимо соблюдение Диеты № 5.
Диетическое ограничение показано при лечении различных наследственных болезней обмена аминокислот/углеводов (непереносимость фруктозы/лактозы, галактоземия, гистидинемия, цистинурия и др.) и других синдромов с известным первичным дефектом. Однако, диетическое ограничение рациона питания должно сопровождаться регулярным биохимическим контролем показателей обмена веществ.
Профилактика
Генетические синдромы у детей разнообразны и некоторые из них протекают тяжело, вызывая раннюю инвалидизацию. Предупредить их можно, проведя генетические исследования лиц, собирающихся стать родителями. После этого проводится медико-генетическое консультирование пары и сообщается о возможном риске рождения больного ребенка. Если этого не было сделано на этапе подготовки к беременности, то для раннего выявления заболевания проводится пренатальная диагностика на ранних сроках. Материалом для исследования служит амниотическая жидкость, биоптаты хориона и ДНК-диагностика.
Немаловажное значение в предупреждении генных мутаций отводится здоровому образу жизни и правильному питанию женщины, готовящейся стать матерью и беременной. В питании должны присутствовать антимутагены — вещества, способные подавлять мутации как спонтанные, так и индуцированные. Это незаменимые аминокислоты, полиненасыщенные жирные кислоты, витамины (А, Е, С, К), пищевые волокна и микроэлементы. Есть вещества, которые связывают мутагены и канцерогены до поступления их в клетку — это пищевые волокна, клетчатка, лигнин, пектин целлюлоза. Полезны в этом плане петрушка, укроп, яблоки, сливы, сельдерей, свекла, отруби. Есть вещества, которые влияют на ферментные системы детоксикации — индолы, полифенолы, флавоноиды, монотерпены и растительные фенеолы. Эти вещества содержат белокочанная капуста, цветная капуста и брюссельская, морковь, сельдерей петрушка, соя, горох, красная фасоль, чечевица, зеленый горошек, цитрусовые, грецкие орехи, фундук, миндаль, фисташки, кедровые орехи, темный виноград.
Вторичная профилактика наследственных заболеваний при дефиците А1АТ включает:
- отказ от курения;
- обязательная вакцинация против пневмококковой инфекции и гриппа, а при поражении печени — против гепатита;
- сведение к минимуму или исключение раздражающих факторов: испарения, пыль, табачный дым (при пассивном курении).
Больным с поражением печени на фоне дефицита А1АТ — отказ от алкогольных напитков и соблюдение Диеты № 5.
Последствия и осложнения
Осложнения при синдроме Нунан:
- гидроцефалия;
- судороги;
- краниосиностоз (раннее закрытие швов черепа);
- аномалия Арнольда-Киари (аномалия головного мозга и различные неврологические нарушения).
Осложнения при синдроме Блума включают:
- хронические заболевания легких;
- сахарный диабет;
- умственная отсталость;
- риск возникновения онкологических заболеваний (лейкоз, карцинома, лимфома, развивающиеся в молодом возрасте).
При синдроме Прадера-Вилли среди осложнений стоит выделить:
- выраженную степень ожирения;
- апноэ во сне;
- сахарный диабет;
- почечную недостаточность;
- риск возникновения онкологических заболеваний (лейкоз).
При дефиците альфа-1-антитрипсина у больных развиваются:
- эмфизема легких;
- идиопатический фиброз;
- рак легких;
- спонтанный пневмоторакс;
- бронхоэктазы;
- цирроз печени.
Прогноз
При диагностировании синдрома Прадера-Вилли на раннем этапе и проведении поддерживающего лечения (диетическая коррекция, массаж, ЛФК, введение гормона роста), прогноз заболевания относительно благоприятный. Продолжительность жизни достигает 60 лет. Смерть наступает в результате осложнений ожирения — сердечная недостаточность, апноэ во сне, декомпенсированный сахарный диабет, патологией почек с почечной недостаточностью.
При синдроме Блума у больных отмечается высокая вероятность онкологии, диабета, обструктивной болезни легких, что сказывается на состоянии здоровья и прогнозе. Раннее начало и быстрое прогрессирование обструктивной болезни уменьшают продолжительность жизни — больные живут не более 28 лет. Причиной смерти становятся также злокачественные новообразования.
Прогноз при дефиците А1АТ зависит от воздействия пыли, курения, обострений бронхита, наличия бронхиальной астмы и эмфиземы. Причина смерти при этой патологии — эмфизема с дыхательной недостаточностью, цирроз и рак печени.
Прогноз синдрома Ваарденбурга чаще всего благоприятный, поскольку пороки развития не прогрессируют и не угрожают жизни пациента. Очень редко аномалии развития в виде порока сердца, гидроцефалии или патологии кишечника могут ухудшить прогноз. Снижения качества жизни вызывает тугоухость и глухота, которые должны своевременно корректироваться, поскольку при поздней диагностике они приводят к нарушению речи.
Список источников
- Пузырев В.П. Феномо-геномные отношения и патогенетика многофакторных заболеваний // Вестн. РАМН. 2011. № 9. С. 17–27.
- Диагностика и лечение легочной патологии при дефиците альфа-1-антитрипсина: доклад Европейского респираторного общества/Пульмонология. 2018.— 28 (3), С. 273–295.
- Семячкина А. Н., Воскобоева Е. Ю., Букина Т. М. Клинико-генетическая характеристика муколипидоза II и IIIA типов у детей/Российский вестник перинатологии и педиатрии. 2017.— 62:(3), С. 71-78.
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Новиков П. В. Лизосомные болезни накопления - актуальная проблема педиатрии и современные возможности патогенетического лечения / Российский вестник перинатологии и педиатрии. - 2014. - №4, С. 4-9.
Комментарии








Статьи по теме





Последние комментарии
Людмила: Гусиная кожа это не заболевание, а состояние кожи, и улучшить его можно увлажнением ...
вера: Сыну было 4 мес, путал день с ночью, укачивала часами, решила попробовать Дормикинд ...
Сергей: Здраствуйте! Подскажите где можно достать этот препарат Денебол гель?
Надежда: Наконец нашла эту статью, где рассказано как правильно питаться, после удаления желудка ...