Синдром Мартина-Белл


- Лечащие врачи: Неонатолог, Педиатр, Невролог, Психиатр, Психотерапевт, Генетик, Логопед
Общие сведения
Умственная отсталость относится к распространенным формам инвалидности и встречается у 13% населения. Одной из причин задержки умственного развития является синдром Мартина-Белл (син. синдром ломкой Х-хромосомы, синдром фрагильной Х-хромосомы, FRAX-А), представляющий собой наследственное заболевание, сочетающее снижение интеллектуального развития, специфические фенотипические особенности, расстройства аутистического спектра и специфические поведенческие характеристики, развитие которых обусловлено фрагильным участком Х-хромосомы в области Xq27. Свое название синдром Мартина-Белл получил в 1943 по фамилиям врачей, впервые описавших патологию.
Синдром Мартина-Белл является второй причиной (после синдрома Дауна) случаев семейной умственной отсталости (аутизм) и развития различных поведенческих проблем (синдром гиперактивности). Форма Х-сцепленной умственной отсталости затрагивает преимущественно лиц мужского пола мужчин, в то время как у женщин-носителей мутации в ряде случаев вследствие инактивации хромосомы с нормальным аллелем может приводить к незначительно выраженной умственной недостаточности. Синдром Мартина-Белл обусловлен истончением хромосомы в определенном участке. Эту хрупкость (ломкость) обнаружили еще в прошлом веке, а позже был выявлен ген FMR1, который отвечает за появление данной «ломкости», развивающейся в результат прекращения выработки нуклеоцитоплазматического белка типа FMR1, который необходим для связывания разнообразных мРНК/осуществления необходимых процессов внутри рибосом и правильного созревания нервной системы.
Частота встречаемости синдрома Мартина-Белл среди мальчиков составляет 1:4000 и 1:6000 среди девочек. При этом, следует учитывать, что наследование/развитие заболевания зависит от пола, поскольку ген FMR1, обеспечивающий секрецию/транспортировку специфических белков и когнитивное развитие расположен на Х-хромосоме, которых у мужчин одна, а у женщин — две. Соответственно, если Х-хромосома оказалась ломкой у мужчин развивается заболевание, в то время как у женщин наличие одной Х-хромосомы с мутантным геном FMR1 может клинически не проявляться или заболевание может развиваться в 30-50% случаев.
Патогенез
В основе патогенеза заболевания лежит многократное увеличение дупликаций единичных тринуклеотидов ЦГГ, расположенных на участке Xq27.3 гена FMR1, ответственного за кодирование белка FMRR, который отвечает за развитие ЦНС (формирование аксонов/синапсов, появление/усложнение нейронных связей, успешность процессов запоминания/обучения).

При наследственном синдроме Мартина-Белл участок хромосом, который подвержен структурным изменениям, характеризуется различным удлинением повторяющихся последовательностей тринуклеотидов ЦГГ и может находиться в различных состояниях:
- количество повторов от 6 до 39 определяется как норма (отсутствии болезни/носительства);
- количество повторов в диапазоне 40-60 повторов является промежуточным состоянием (заболевание отсутствует);
- количество повторов в диапазоне 55-200 диагностируется как состояние премутации;
- количество повторов в диапазоне более 200 (чаще от 230 до 4 000) определяется как полная мутация, что приводит к полному подавлению транскрипции и недостаточному производству белка FMR1.
Классификация
При синдроме ломкой Х-хромосомы мутация гена существует в двух формах, имеющих отличия по своей пенетрантности:
- стадия премутации (не имеющая фенотипических проявлений);
- стадия полной мутации, развивающаяся при прохождении через женский мейоз (появление аномалий).
Причины
Причиной развития синдрома ломкой х-хромосомы является значительное увеличение количества CGG-повторов (более 200) в гене FMR1 (Xq27.3), что приводит к нарушению синтеза белка FMRR. Этот белок участвует в регуляции экспрессии/трансляции множества различных видов мРНК, кодирующих белки, принимающих участие в развитии/поддержании системы синаптических нейрональных связей. Следует учитывать, что число повторов в диапазоне 60-200 (премутация) сопровождается высоким риском увеличения повторов в последующем поколении и, соответственно, развитием заболевания.
Синдром Мартина-Белл относится к группе заболеваний, сцепленных с полом (Х-хромосомой). Наследование синдрома Мартина-Белл происходит по доминантному типу с неполной пенетрантностью. Поскольку у мужчин присутствует лишь одна X-хромосома, мутантный аллель провоцирует болезнь всегда. У женщин имеются две половые X-хромосомы, одна из них активная, а другая – инактивированная. Соответственно заболевание у женщин при наличии мутации в одном из любых двух генов FMR1 может отсутствовать. Мужчины с ломкой хромосомой X передать ее сыновьям не могут, однако, передают всем дочерям, которые могут оставаться здоровыми носителями мутации или болеют. В тоже время женщины с дефектной хромосомой с вероятностью 50% передают ее детям обоих полов. При этом, наследование синдрома от поколения к поколению учащается (парадокс Шермана).

Симптомы
Основным проявлением синдрома Мартина-Белл у мальчиков является умственная отсталость, проявляющаяся выраженным снижением интеллекта и недостаточностью развития сложных форм логического/абстрактного мышления и памяти. Такие пациенты не понимают абстрактно-логические высказывания/предложения, не могут проводить аналогии и обобщать информацию. Словарный запас крайне беден. Анализ, сравнение и обобщение доступны лишь на примитивном уровне, в частности, в конкретных бытовых ситуациях. У большинства пациентов IQ варьирует в пределах 40–50 баллов, в редких случаях достигает 70–80. Зрительное восприятие/номинативная речь сохранена. У девочек снижение когнитивных функций менее выраженное, и в целом, соответствует пограничному уровню интеллектуального развития, реже – легкой степени олигофрении. Также к типичным проявлениям заболевания относится специфика речи, что проявляется ее ускорением, сбивчивостью, повторами, навязчивостью повторений какой-либо фразы.
Аутистические расстройства проявляются трудностями в коммуникации как в семье, так и в социуме и поведенческими нарушениями. Такие дети зачастую замкнуты, не входят в зрительный контакт, избегают социума и агрессивны при попытке установления с ними контакта. В более тяжелых случаях отмечается полное отсутствие речи как средства общения (мутизм).
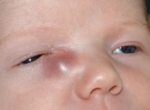
К характерным проявлениям синдрома Мартина-Белл относятся фенотипические особенности ребенка: выступающий широкий лоб и подбородок, клювовидно загнутый нос, большие низко расположенные уши, вытянутое лицо с уплощенной средней частью, деформация грудной клетки, увеличение яичек, высокое сводчатое нёбо. Отмечается чрезмерная подвижность суставов, повышенная масса тела, пролапс митрального клапана, гиперэластичность кожи, радужные оболочки глаз чаще светлого оттенка. При этом выраженность фенотипических признаков может существенно варьировать от едва определяемых одного-двух признаков до полного комплекса. Фото синдрома Мартина-Белл приведено ниже.

В поведении ребенка преобладает гиперактивность, двигательная расторможенность, стереотипии, склонность к самоповреждениям. Стереотипные движения проявляются прыжками, хлопками/встряхиваниями руками, вращения вокруг оси, руками, бегом по кругу, хныканьем, гримасничаньем. Существуют трудности контроля поведения, пространственной координации, планирования и переключения внимания. Пациенты не допускают прикосновений и избегают смотреть в глаза, однако интерес к общению присутствует по сравнению с аутизмом.
Специфических неврологических симптомов нет. Присутствует двигательная дискоординация/незначительное снижение мышечного тонуса. Характерно слабое развитие мелкой моторики, что затрудняет освоение игровых/бытовых навыков (рисование, шитье), письма. Реже наблюдаются усиление сухожильных рефлексов, паракинезы, глазодвигательные/экстрапирамидные нарушения (гримасничанье, нахмуривание бровей, зажмуривание глаз). У женщин с премутационным состоянием зачастую развивается первичная недостаточность яичников. Почти у трети пациентов, преимущественно у мужчин, развивается тремор/мозжечковая атаксия (нарушение равновесия), деменция.
Анализы и диагностика
Постановка диагноза при наличии выраженных фенотипических изменений (характерные особенности лица/увеличенные размеры яичек), а также снижение интеллекта, нарушение коммуникации, отставание умственного развития, поведенческие/речевые отклонения усиленные сухожильные рефлексы/мышечный гипотонус, выявляемые при клиническом осмотре и оценки психического статуса ребенка позволяют установить предварительный диагноз. Для окончательного диагноза требуется проведение генетического исследования, сутью которого является изучение строения ДНК цитогенетическим методом и ПЦР: проводится определение количество повторов ЦГГ, а также статус метилирования. Диагноз ставится при количестве триплетных повторов более 200. При 60-199 повторах (если показатель диагностирован у женщины) присутствует риск развития невыраженных фенотипических проявлений и высокий риск развития патологии у детей в следующем поколении. С помощью ПЦР анализируется состав/структура тринуклеотидов.
Также проводится генеалогический анализ (наличие родственников, особенно по мужской линии с данным заболеванием). В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники, чаще – мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
Дифференциальная диагностика проводится с ранним детским аутизмом и заболеваниями, сопровождающимися умственной отсталостью, не обусловленной ломкостью Х хромосомы.
Лечение
В основе лечения синдрома Мартина-Белл лежат методы психолого-педагогической коррекции и симптоматическое медикаментозное лечение. Коррекция направлена на минимизацию отклонений в эмоционально-поведенческой сфере, овладение навыками речи, чтения/письма, ходьбы и общения. Для социализации ребенка используются индивидуальные/групповые тренинги, а также методы когнитивно-поведенческой терапии.
Медикаментозная терапия подбирается в зависимости от симптоматики и включает прием нейролептиков (после 3-х лет) — Галоперидол, Хлорпромазин, Перициазин, доза приёма которых рассчитывается в зависимости от массы тела ребенка. Также для лечения синдрома Мартина-Белл могут назначаться препараты мозга животных (Церебролизин, Церебролизат), действующими веществами которых являются пептиды, способствующие выработке в нейронах белка, пополняя их недостаток.
При возбуждении показаны препараты с седативным действием — Седуксен, Диазепам. Медикаментозная терапия включает назначение психостимуляторов, антидепрессантов (Кломипрамин, Флувоксамин, Сертралин, Флуоксегин, Амитриптилин, Коаксил, Дулоксетин); цитомединов (Кортексин, Солкосерил, Лидаза); антиконвульсантов (Прегабалин, Габапентин), нейрометаболиков (Актовегин, Нейробион), миорелаксантов (Мидокалм, Сирдалуд, Толперизон, Баклофен), ноотропов (Фенибут, Пирацетам, Пантогам, Целестаб, Новобенон, Нооцил, Нейропол и др.).
При первичной недостаточности яичников могут назначаться гормональные препараты (эстрогены — эстрадиол-валерат, эстриол, этинилэстрадиол, эстрофем, овестин, климару, прогестерон, левоноргестрел, норгестрел, прегнил, туринал и др.). По показаниям применяют и другие лекарственные препараты.
Доктора



Лекарства
 Галоперидол
Галоперидол Церебролизин
Церебролизин Церебролизат
Церебролизат Седуксен
Седуксен Диазепам
Диазепам Амитриптилин
Амитриптилин Коаксил
Коаксил Кортексин
Кортексин Солкосерил
Солкосерил Лидаза
Лидаза Актовегин
Актовегин Нейробион
Нейробион Пирацетам
Пирацетам Пантогам
ПантогамГалоперидол, Хлорпромазин, Перициазин, Церебролизин, Церебролизат, Седуксен, Диазепам, Кломипрамин, Флувоксамин, Сертралин, Флюоксегин, Амитриптилин, Коаксил, Дулоксетин, Кортексин, Солкосерил, Лидаза, Прегабалин, Габапентин, Актовегин, Нейробион, Мидокалм, Сирдалуд, Толперизон, Баклофен, Фенибут, Пирацетам, Пантогам, Целестаб, Новобенон, Нооцил, Нейропол.
Процедуры и операции
При синдроме Белл широко используется упражнения в бассейне, ЛФЛ, гирудотерапия, душ Шарко, иглоукалывание, массаж, грязевые ванны с радоном.
Хирургическое вмешательство при осложнениях синдрома Мартина-Белл, затрагивающих жизненно важные органы (врожденные пороки сердца, киста яичников с возможностью трансформации в злокачественное образование). Также хирургические методы лечения (пластическая коррекция) используются для устранение физических дефектов — изменение формы ушей, аномалии половых органов и др.
Диета
Специальной диеты при синдроме Мартина-Белл нет. Рацион питания строится на основе принципов рационального питания в соответствии с возрастом, полом, уровнем физической нагрузки.
Профилактика
На сегодняшний день единственным методом профилактики является пренатальный скрининг беременных на стадии ведения беременности/планирования семьи, что позволяет прервать беременность на раннем этапе при выявлении патологии в Х-хромосоме или определить риск рождения больного ребенка. В качестве альтернативы может быть рекомендовано ЭКО с использованием донорских ооцитов. Генетический тест, позволяющий определить количество повторов в гене FMR1 необходимо пройти:
- женщинам с синдромом преждевременного истощения яичников;
- пациентам/и их родственникам с интеллектуальной недостаточностью;
- лицам, в семье которых были случаи синдрома ломкой Х-хромосомы;
- женщинам с нарушениями фертильности/репродуктивными проблемами, обусловленными повышенным уровнем ФСГ (фолликулостимулирующего гормона);
- пациентам с мозжечковой атаксией/поздно проявившимся тремором.
Последствия и осложнения
К основным осложнениям синдрома Мартина-Белл относятся пороки сердца, а также кистозное перерождение яичников с возможностью трансформации в злокачественное образование.
Прогноз
Ломкость X-хромосомы, если она не осложнена другими патологиями внутриутробного развития плода, имеет благоприятный прогноз для жизни и неблагоприятный для выздоровления. Продолжительность жизни пациента с синдромом ломкой X-хромосомы укладывается в парадигму здоровых людей. При адекватной психологической коррекции/постоянном симптоматическом лечении, помощи больному с адаптацией в социуме качество жизни пациента нормализуется. В ряде случаев синдром Мартина-Белл приводит к инвалидизации.
Список источников
- Зеленова М.А., Юров Ю.Б., Ворсанова С.Г., Юров И.Ю. Психологические аспекты генетических синдромов, ассоциированных с аутизмом и умственной отсталостью // Международный журнал прикладных и фундаментальных исследований. 2015. № 12(10). C. 1870-1876.
- Кузнецова Е.Б., Стрельников В.В., Танас А.С. и др. Технология комплексной ДНК-диагностики синдрома ломкой Х-хромосомы // Медицинская генетика. 2018. Т. 17, № 6. С. 18-23.
- Малов А.Г. Нейропсихологические расстройства при синдроме ломкой х-хромосомы/ журнал Социальные и гуманитарные науки: теория и практика 2019. С. 668-674.
- Тюшкевич С.А. Особенности поведения и когнитивных нарушений у детей и подростков с синдромом умственной отсталости, сцепленной с ломкой хромосомой Х: автореф. дис. … канд. психол. наук. М., 2010. 27 с.
- Новиков П.В., Баранов А.А., Каганов Б.С., Шиляев Р.Р. Руководство по педиатрии. Врожденные и наследственные заболевания. Издательский Дом «Династия», 2007. - 544 с.
Комментарии







Последние комментарии
вера: Сыну было 4 мес, путал день с ночью, укачивала часами, решила попробовать Дормикинд ...
Сергей: Здраствуйте! Подскажите где можно достать этот препарат Денебол гель?
Надежда: Наконец нашла эту статью, где рассказано как правильно питаться, после удаления желудка ...
Алла Анатольевна: Как только прочитаешь противопоказания, сразу сдохнуть хочется.