Митохондриальные заболевания
Общие сведения
Митохондриальные болезни человека – это генетически обусловленные патологии, которые возникают из-за нарушения функций и структуры митохондрия. Они развиваются вследствие наследственных мутаций в митохондриальной ДНК или в ядерных генах. Известно порядка 300 таких мутаций, ежегодно появляются новые. Врачам не всегда удается определить, что стало причиной митохондриального заболевания. Большая часть пациентов имеет диагноз без подтвержденного генетического дефекта.
Зачастую митохондриальные заболевания связаны с повреждением электрон-транспортной цепи митохондрий и нарушением синтеза аденозинтрифосфорной кислоты (АТФ). Из-за этого их еще называют патологиями дыхательной цепи митохондрий или болезнями окислительного фосфорилирования.
Митохондриальные заболевания приводят к нарушению тканевого дыхания, поскольку характеризуются дефектами митохондрий – микроскопических органелл, производящих до 95 % всей клеточной энергии.

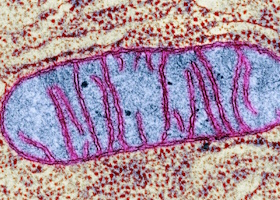
Строение митохондрия
Митохондрии отвечают за клеточное дыхание, рост и развитие клеток, участвуют во всех видах обмена. Основная их функция – это преобразование питательных веществ в ходе синтеза АТФ. В каждой клетке находится несколько сотен или тысяч митохондрий, они занимают порядка 10-20% ее объема.

Большая их часть находится в органах, которые сильно нуждаются в энергии. Это головной мозг, сердце, мышцы и жировая ткань, которые больше всего страдают при митохондриальных заболеваниях. В зависимости от вида пораженных клеток, могут появляться следующие проблемы со здоровьем:
- слабость глазных мышц;
- эпилептические судороги;
- снижение мышечного тонуса;
- кардиомиопатия;
- нарушение слуха или зрения;
- патологическая утомляемость;
- нейтропения;
- задержка полового созревания и т. д.
С развитием митохондриальной медицины стало известно, что дисфункция митохондрий влияет на возрастные изменения и развитие болезней, трудно поддающихся лечению. Прежде всего речь идет об онкологии, эндокринных нарушениях, заболеваниях сердца и нервной системы. В последние годы была доказана взаимосвязь между функционированием митохондрий и возраст-ассоциированными болезнями, к которым относится:
- болезнь Альцгеймера и Паркинсона;
- сахарный диабет;
- ожирение;
- инфаркт миокарда;
- шизофрения.
В отличие от других внутриклеточных структур, митохондрии имеют часть собственного генетического материала, который наследуется только по материнской линии. Несмотря на то, что мать передает мутированный ДНК всем своим детям, симптомы не обязательно будут у всех. Клинические проявления и их тяжесть могут существенно отличаться у разных членов семьи.
Митохондриальное заболевание может дебютировать в любом возрасте. Чем позже оно проявится, тем благоприятнее прогноз. В большинстве случаев патология развивается в детском возрасте. Начинающиеся внутриутробно болезни зачастую приводят к порокам головного мозга и имеют летальный исход.
Пути наследования
Нарушения митохондрий связаны с большим количеством заболеваний, которые могут быть как врожденными, так и приобретенными, иметь метаболическое, воспалительное, дегенеративное происхождение. Однако в основе патогенеза находятся генетические мутации, которые могут наследоваться по следующим типам:
- Материнский. Это классический тип наследования, присущий большинству митохондриальным заболеваниям у детей. Дефектная ДНК наследуется от матери. Может передаваться детям обеих полов. У матери не обязательно будут симптомы заболевания. Материнский митохондрий имеет собственную ДНК, поэтому больной мужчина не сможет передать мутацию ребенку.
- Аутосомно-рецессивный. Вероятность генетической мутации у ребенка при таком типе наследования составляет около 25 %. Оба родителя могут быть носителями дефектного гена, однако сами не имеют симптомов, поскольку у них есть копия здорового ДНК. Клинические симптомы появляются в случае, когда ребенок получает дефектный ген от матери и отца одновременно. Если ему передается только одна копия аномального гена, то он становится здоровым носителем. Зачастую болеет только одно поколение.
- Аутосомно-доминантный. Редкий тип наследования, при котором достаточно одной копии дефектного гена, чтобы развилась болезнь. В каждом поколении есть случаи митохондриальных заболеваний, болеют оба пола.
- X-сцепленный. Генетические мутации передаются от матерей сыновьям. Женщины являются бессимптомными носителями, имеют одну дефектную и одну нормальную Х-хромосому. Наиболее часто по такому типу наследуется недостаточность пируватдегидрогеназы.
Симптомы
Медицине известно порядка 150 видов митохондриальных патологий, поэтому клинические проявления разнообразные. Симптоматика зависит от органа-мишени:
- Нервная система. Мигрень, судороги, поражение периферических нервов, деменция, атаксия, инсульт, интенционный тремор, инсультоподобные пароксизмы.
- Сердце. Аритмия, кардиомиопатия, нарушение проводимости.
- Мышцы. Непереносимость физических нагрузок, гипотония, миопатия, повышенная утомляемость.
- Поджелудочная железа. Сахарный диабет.
- Почки. Гломерулонефрит, синдром Фанкони.
- Органы зрения. Птоз, косоглазие, ухудшение или потеря зрения.
- Органы слуха. Ухудшение или потеря слуха.
- Печень. Печеночная недостаточность и другие заболевания.
- Кровеносная система. Анемия, нейтропения, тромбоцитопения.
- ЖКТ. Запор, диарея, недобор веса, нарушение всасывания питательных веществ, непроходимость кишечника.
Первыми зачастую страдают мышцы. В большинстве случаев также поражается нервная система, возможны дисметаболические (рвота, летаргический сон, кома) и дыхательные нарушения по типу апноэ, тахипноэ, гипервентиляции.
Митохондриальным заболеваниям присуще прогрессирующее течение. При длительном существовании нарушается работа печени, почек, поджелудочной железы и других внутренних органов.
Диагностика
Постановка диагноза может быть затруднена, занимать много времени. Зачастую, прежде чем сделать верное заключение, доктору приходится исключить ряд патологий, среди которых:
- лейциноз;
- тирозинемия;
- орнитинтранскарбамилазная недостаточность;
- синдром Прадера-Вилли;
- ДЦП;
- болезнь Помпе;
- диабет;
- гипопаратиреоз и другие.
Для дифференциальной диагностики используют исследования, позволяющие выявить биохимические и нейрофизиологические маркеры митохондриальных заболеваний. Отличить патологию, спровоцированную дисфункцией митохондрия, от болезни обмена веществ можно по наличию лактоацидоза.
Пациенту могут назначить следующие методы диагностики:
- биохимический анализ крови;
- цитоморфоденситометрические исследования;
- методы нейровизуализации;
- ультразвуковые исследования внутренних органов;
- биопсия скелетных мышц с гистохимическим исследованием.
Клиническими критериями митохондриальных заболеваний может стать:
- отягощенный семейный анамнез;
- внезапное ухудшение состояние здоровья ребенка, не поддающееся традиционному лечению, например, сниженный тонус мышц, судороги, рвота, слабость, расстройство дыхания, поражение почек, печени;
- отставание в умственном развитии без причины;
- признаки поражения соединительной ткани;
- нейродегенеративные симптомы.
При ярко выраженной картине какого-либо митохондриального синдрома доктор назначает генетическое обследование, результаты которого и являются основанием для постановки верного диагноза.
Примеры митохондриальных заболеваний
Все митохондриальные болезни можно подразделить на 2 группы:
- Первичные. Возникают из-за повреждения митохондриальной ДНК и нарушения процесса окислительного фосфорилирования. Примером является оптическая нейропатия Лебера, синдром Кернса-Сейра, MELAS, MERRF и другие.
- Вторичные. Спровоцированы мутациями ядерной ДНК и нарушением структуры белков, влияющих на процесс окислительного фосфорилирования. К этой группе относится атаксия Фридрейха, болезнь Вильсона, Хантингтона, Паркинсона, митохондриальная энцефаломиопатия и другие.
Одни и те же генетические мутации могут вызывать разные заболевания, отличающиеся клиническими проявлениями. Это генокопии. Их пример — болезнь Альперса, синдром SANDO. Бывает и обратное, что мутации в разных генах приводят к одинаковым болезнями. Такой феномен в генетике называют фенокопии.
К основным митохондриальным патологиям относится:
- Оптическая нейропатия или атрофия зрительного нерва Лебера. Характеризуется повреждением ганглиозных клеток сетчатки глаза. Сопровождается прогрессирующей потерей зрения, зачастую в возрасте 18-30 лет. В некоторых случаях симптоматика дополняется проблемами с сердцем, мышечной слабостью и спазмами. Болезнь передается детям по материнской линии.
- Синдром Ли или подострая некротизирующая энцефаломиелопатия. Заболевание дебютирует в период с 3 месяцев до 2 лет, крайне редко возникает в подростковом возрасте. Проявляется отставанием в развитии, плаксивостью, раздражительностью, рвотой, судорожными припадками, мышечной гипотонией, лактоацидозом.
- Синдром MELAS, включающий в себя митохондриальную энцефаломиопатию, лактоацидоз и инсультподобные пароксизмы. Характеризуется поражением центральной нервной системы (ЦНС), мышц и отдельных внутренних органов. Клиническая картина включает эпилептические приступы, симптомы инсульта, мышечную слабость, непереносимость физических нагрузок. Лечение предполагает прием метаболических препаратов и симптоматическую терапию.
- Синдром Кернса-Сейра. Это митохондриальная миопатия, которая дебютирует в возрасте до 20 лет. Характеризуется поражением мышц, участвующих в движении век и глаз, двусторонней пигментной ретинопатией и блокадой сердца. Клиника синдрома — птоз, медленно прогрессирующая офтальмоплегия, атрофия зрительного нерва, пигментный ретинит, снижение остроты зрения.
- Синдром MERRF или миоклонус-эпилепсия с рваными красными волокнами. Характеризуется тяжелым поражением ЦНС и мышечной ткани. Клиническая картина включает нарушение координации движений, мышечную слабость, ноющую боль в икроножных мышцах, неустойчивость при ходьбе, эпилептические приступы. Диагноз подтверждается посредством ДНК-анализа и гистологического исследования мышечных волокон, полученных в ходе биопсии.
- Митохондриальная нейрогастроинтестинальная энцефалопатия (MINGIE). Прогрессирующее аутосомно-рецессивное заболевание, приводящее к поражению тканей нервной и пищеварительной системы. Ранняя диагностика и вовремя начатое лечение позволяет отсрочить летальные осложнения. Патология проявляется нарушением моторики кишечника, полинейропатией, лейкоэнцефалопатией, птозом, нейросенсорной глухотой. Дебют приходится на возраст до 20 лет.
- Синдром SANDO. Фенотип, ассоциированный с мутациями в генах POLG или TWNK. Может наследоваться по аутосомно-доминантному или аутосомно-рецессивному типу. Характеризуется нарушением координации движений и равновесия, поражением нервов, дизартрией и снижением подвижности глазных яблок.
- Синдром MEMSA. Первый симптом болезни — это церебральная атаксия, далее возникает миопатия и миоклоническая эпилепсия.
- Синдром MIRAS. В детском возрасте развивается по типу энцефалопатии, во взрослом – атаксии. Клинически проявляется нарушением координации движений, когнитивными расстройствами, изменениями личности, нейропатией, эпилепсией.
- Синдром NARP. Характеризуется атаксией, нейропатией и пигментным ретинитом, порой случаются эпилептические приступы.
- Синдром Вольфрама. Это прогрессирующее нейродегенеративное заболевание, сопровождающееся неаутоиммунным инсулинзависимым сахарным диабетом.
- Синдром Барта. Характеризуется поражением сердца, скелетной мускулатуры и системы крови. Основу клинической картины составляет нейтропения, кардиомиопатия и скелетная миопатия.
- Болезнь Альперса-Хуттенлохера. Триада симптомов — прогрессирующая регрессия развития, тяжелые судороги и печеночная недостаточность. Болезнь неуклонно прогрессирует и без врачебной помощи приводит к летальному исходу.
- Атаксия Фридрейха. Развивается до 25 лет. Сопровождается поражением центральной и периферической нервной системы, клеток сердца, сетчатки, костей, поджелудочной железы. Первые признаки болезни — неврологические нарушения. К ним постепенно присоединяется слабость мышц, нарушение речи, слуха, зрения. В тяжелых случаях пациент теряет все сухожильные и надкостничные рефлексы, атрофируются верхние конечности, появляются тазовые нарушения, развивается слабоумие.
Осложнения
Самым тяжелым осложнением митохондриальных заболеваний становится поражение ЦНС. Отек головного мозга, эпилептические приступы, судороги могут стать причиной летального исхода. Не меньшую угрозу несут миопатии, поскольку слабость мускулатуры дыхательной системы также может стать причиной смерти пациента.
При митохондриальных заболеваниях страдает двигательная функция, ухудшается зрение, слух, когнитивные способности, может развиваться гломерулонефрит, сахарный диабет, хронический панкреатит.
Лечение и прогноз
Большая часть митохондриальных заболеваний — это тяжелые прогрессирующие патологии, от которых не существует специфического лечения. Терапия направлена на купирование существующих симптомов.
Продолжительность и качество жизни зависит от характера течения заболевания, вида генетической мутации. Неблагоприятный прогноз имеют манифестные формы болезней, характеризующиеся клинически выраженной симптоматикой.
Список источников
- Оптическая нейропатия Лебера/ Е. А. Руина, О. И. Чадаева, Е. В. Паршина, А. А. Смирнов// Медицинский альманах. – 2017. – №5.
- Millar W.S., Lignelli A., Hirano M. MRI of Five Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy. AJR 2004;182:1537 – 41
- Наследственные болезни нервной системы: Рук. для врачей/ Под ред. Ю. Е. Вельтищева, П. А. Темина. - 1998.
- Наследственные болезни обмена веществ/ Краснопольская К.Д. - 2005.
- Митохондриальная энцефалопатия с инсультоподобными эпизодами и лактат-ацидозом (синдром MELAS): критерии диагностики, особенности эпилептических приступов и подходы к лечению на примере клинического случая/ Ямин М. А., Черникова И. В., Арасланова Л.В., Шевкун П.А.// Неврология, нейропсихиатрия, психосоматика – 2017 – №9.

Комментарии




Похожие статьи







Последние комментарии
essam: Что-то меня дернуло и решила я попробовать новый гель для интимной гигиены. Баночка такая ...
essam: Несколько месяцев назад, спустя почти 2 года свободы, у меня появились отношения. Вместе ...
Инна: Что-то меня дернуло и решила я попробовать новый гель для интимной гигиены. Баночка такая ...
Вероника: Все понятно,но действительно что при ПН можно например рис,компот,мед при диабете ...