Липодистрофия и болезнь Уиппла

- Лечащие врачи: Эндокринолог
- Диеты при болезни: Диета 10-й стол, Диета при повышенном сахаре в крови, Гипохолестериновая диета, Стандартная гиполипидемическая диета
Общие сведения
Сегодня мы рассмотрим некоторые врожденные и приобретенные заболевания, которые связаны с нарушением обмена жиров. Термин липодистрофия (синоним жировая дистрофия) — объединяет редкие наследственные или приобретенные заболевания, при которых отмечается полная (генерализованная форма) или частичная (парциальная, локальная форма) потеря жировой клетчатки. Также отмечается перераспределение избытка жировой клетчатки (липогипертрофия) в области шеи, плеч, над ключицами, живота. При любой форме не столько важны внешние косметические дефекты, сколько различные метаболические нарушения и изменения в важных органах, приводящие к нарушению их функции.
Основная причина смерти больных при различных формах липодистрофий — метаболические нарушения (сахарный диабет, жировой гепатоз, инсулинорезистентность, дислипидемия, мочекаменная болезнь), кардиомиопатия, инфаркт миокарда, различные виды аритмий, гепатоцеллюлярная карцинома, панкреатит, почечная недостаточность. Поэтому больным рекомендуется ежегодные обследования на предмет сахарного диабета, заболеваний печени, почек и сердечно-сосудистой системы. Важно выявлять единичные и семейные случаи заболеваний проведением молекулярно-генетической диагностики.
Болезнь Уиппла (синоним кишечная или интестинальная липодистрофия) и липодистрофии объединяет только термин и нарушение метаболизма липидов, но причины и механизм развития заболеваний, лечение абсолютно разное. Болезнь Уиппла — это редкое системное инфекционное заболевание, которое вызывается бациллой Tropheryma whippelii. При этом заболевании изначально поражаются тонкая кишка и лимфатические узлы брюшной полости, в которых отмечается диффузное отложение жира, а местами образуются жиросодержащие полости.
Патолог, описавший это заболевание, считал причиной его развития именно нарушение обмена липидов и им был предложен термин «интестинальная липодистрофия». Намного позже выяснилось, что заболевание вызывает Tropheryma whippelii, палочкообразные тельца которой нашли в цитоплазме макрофагов. Поскольку была подтверждена бактериальная природа заболевания, эффективным оказалось длительное антибактериальное лечение. Кроме кишечника в процесс вовлекаются различные органы (при внекишечных формах), то есть заболевание имеет системный характер. При неправильной диагностике и лечении заболевание протекает с осложнениями и может закончиться летальным исходом. Диагностика болезни Уиппла часто вызывает трудности, что связано с разнообразием клинических проявлений. Часто диагноз устанавливается только через 6 лет от начала заболевания.
Патогенез
Патогенез наследственных липодистрофий связан с нарушением образования жировой ткани и ее дифференциации на разных уровнях. Эти нарушения связаны с генетическими мутациями, которые в итоге приводят к атрофии жировых депо и нарушению всего липидного обмена в организме.
Жировую ткань рассматривают как самостоятельный эндокринный орган, который секретирует гормоноподобные вещества — адипонектин, лептин, резистин и другие. Большинство из этих веществ участвуют в воспалительных процессах при ожирении и поддерживают инсулинорезистентность. Уровень адипонектина и лептина снижается при липодистрофии. Считается, что дефицит лептина вызывает уменьшение подкожной клетчатки, усиливает аппетит, способствует неправильному перераспределению и отложению жира, что в свою очередь влечет инсулинорезистентность, сахарный диабет, повышение триглицеридов и ранний атеросклероз, жировую дистрофию печени, поликистоз яичников, черный акантоз.
При врожденной генерализованной липодистрофии у больных затрудняется усвоение липидов, поэтому их уровень в крови повышается и усиливается нагрузка на органы, которые участвуют в их метаболизме. В этих органах (прежде всего печень) происходит аномальное накопление жира и развивается жировой гепатоз, а в последствии стеатогепатит. Жировая дистрофия отмечается в почках, миокарде, нервной ткани, что приводит нарушению их функций.
Болезнь Уиппла характеризуется изменениями в тонком кишечнике, лимфатических узлах брюшной полости и лимфатических сосудах, которые обусловлены действием бациллы Tropheryma Whippelii. Бациллы T. whipplei находят в слизистой оболочке и в макрофагах больных. Макрофаги теряют способность лизировать бациллы, а накопление и размножение их внутри клеток вызывает гибель и скопление жира. Собственный слой слизистой кишечника в большом количестве пропитан специфическими PAS-позитивными макрофагами, которые нарушают строение ворсинок. В слизистой обнаруживают скопления жира, лимфатические сосуды стенки кишечника расширяются, сладки кишечника отекают, а ворсинки изменяются. Эти изменения нарушают всасывание и транспортировку питательных веществ из кишечника в лимфатическое пространство и сосуды слизистой. В патологический процесс также может вовлечься толстая кишка, печень, ЦНС, сердце, надпочечники, синовиальные оболочки, легкие и костный мозг. В патогенезе заболевания большое значение имеют иммунологические нарушения: снижение Т-клеточного иммунитета, уменьшение продукции γ-интерферона и интерлейкина-12.
Классификация
По этиологии липодистрофия бывает:
- Наследственной (связана с мутациями генов, которые передаются по наследству).
- Приобретенной.
При врожденной генерализованной форме у больных от рождения (или в первый год) есть признаки заболевания. При приобретенных признаки отсутствуют до 5–7 лет или подросткового возраста.
По степени утраты жировой ткани:
- Генерализованная (характерно повсеместное отсутствие жирового слоя, затруднение усвоения липидов и накопление их в органах).
- Парциальная (локальное отсутствие жировой клетчатки).
Наследственные формы делятся на несколько групп:
- Врожденные генерализованные.
- Семейные парциальные.
- Липодистрофии при прогероидных синдромах, для которых характерно преждевременное старение.
Врожденные генерализованные липодистрофии
Это большая группа различных наследственных заболеваний. При этом их объединяет нарушение метаболизма жировой ткани, что сопровождается ее уменьшением или полным отсутствием (атрофия). Подкожная жировая клетчатка у больных исчезает на большей части тела, характерны сахарный диабет и черный акантоз (пигментная дистрофия, провоцируемая инсулинорезистентностью и ожирением) кожных складок. Некоторые формы сопровождаются олигофренией разной степени.
В зависимости от мутаций выделяют несколько типов этой врожденной патологии:
- Врожденная липодистрофия типа 1. Она связана с мутацией гена AGPAT2 на 9 хромосоме. При этом типе подкожной клетчаткой не усваиваются липиды, а мембраны жировых клеток из-за дефицита фосфолипидов разрушаются. В течение некоторого времени развивается атрофия жировой ткани, повышаются триглицериды в крови и липиды откладываются в других органах. Нарушение углеводного обмена проявляется сахарным диабетом 2-го типа.
- Заболевание 2-го типа связано с мутацией гена BSCL2 11-й хромосомы. На самых ранних этапах затруднено формирование подкожной клетчатки. Характерна умственная отсталость.
Врожденную липодистрофию типа 3 вызывает мутация гена CAV1 на 7-й хромосоме. Эта мутация приводит к нарушению метаболизма жировой ткани в подкожной клетчатке и в костном мозге, а это влияет на систему гемопоэза. - Врожденную липодистрофию типа 4 связывают с мутацией гена PTRF на 17-й хромосоме. При этой форме нарушается строение и функция адипоцитов (жировых клеток), а также скелетной мускулатуры и клеток миокарда. Поэтому развиваются мышечные нарушения и тяжелая кардиомиопатия с аритмиями и сердечной недостаточностью. У больных с этой формой врожденной липодистрофии часты случаи летального исхода на этом фоне.
Семейные парциальные формы отличаются началом заболевания в детском и подростковом возрасте. В определенных областях утрачивается жировая клетчатка, но отмечается накопление ее в других анатомических областях. Рано развивается инсулинорезистентность, дислипидемия, жировая дистрофия печени, увеличивается печень и селезенка. В крови снижен уровень лептина.
Приобретенные липодистрофии
Эти формы нарушения липидного обмена приобретаются в течение жизни и не передаются по наследству. Они не имеют генетической причины, но при локальной приобретенной форме обнаруживается мутация в гене LMNB и PPARG.
Приобретенное нарушение обмена липидов связано со многими факторами. В связи с этим выделяют жировую дистрофию:
- Связанную с аутоиммунными заболеваниями (системная склеродермия, красная волчанка).
- Развивающуюся при проведении антиретровирусной терапии. Это самая распространенная форма, возникающая у ВИЧ-инфицированных на фоне длительного лечения ингибиторами протеаз.
- Связанную с панникулитом (воспаление жировой клетчатки).
- Связанную с гломерулонефритом.
- Синдром Симонса.
- Медикаментозную (инъекции инсулина, вакцин).
Приобретенная парциальная форма с уменьшением жировой клетчатки на лице, шее и грудной клетке и увеличенным объемом в нижней половине туловища (область бедер, ног и таза) описана врачом Симонсом. Заболевание встречается у женщин и манифестирует при достижении половой зрелости. Заболевание может протекать без нейроэндокринных нарушений — беспокоят только косметические недостатки. Однако, чаще встречается вариант с вестибулярными расстройствами и эндокринными нарушениями: несахарный диабет, сахарный диабет, приступы гипогликемии, нарушение менструального цикла, отеки.
Все липодистрофии (наследственные и приобретенные) сопровождаются различными метаболическими нарушениями — сахарный диабет, инсулинорезистентность, черный акантоз, дислипидемия, синдром поликистозных яичников и стеатогепатит. Выраженность метаболических нарушений зависит от степени утраты жировой ткани, поэтому чаще они встречаются при генерализованных формах.
В результате нарушения усваивания жиров их уровень в крови повышается и, соответственно, повышается нагрузка на органы, которые участвуют в их метаболизме. В норме в цитоплазме клеток присутствуют нейтральные жиры, но в данном случае происходит избыточное накопление жира в клетках — развивается паренхиматозная жировая дистрофия. Прежде всего это касается печени, почек, миокарда, селезенки, нервной ткани.
Паренхиматозная жировая дистрофия печени сопровождается накоплением преимущественно триглицеридов. Сначала в гепатоцитах появляются гранулы жиров (пылевидное ожирение), потом мелкие капли, сливающиеся в крупные (крупнокапельное ожирение). Может быть одна жировая вакуоль, заполняющая всю клетку и отодвигающая ядро к периферии. Гепатоциты становятся похожими на жировые клетки. Стеатоз печени начинается с периферии долек, а при прогрессировании приобретает диффузный характер, что сказывается на функции печени. Стеатоз переходит в стеатогепатит, который заканчивается циррозом печени.
Жировая дистрофия миокарда характеризуется накоплением триглицеридов в клетках миокарда. Процесс носит очаговый характер — изменения в миоцитах происходят в клетках, расположенных по ходу мельчайших вен. При выраженном процессе миокард становится дряблым, сердце увеличивается, камеры растягиваются, появляется исчерченность внутренней оболочки сердца, которое описывается как «тигровое сердце». Выраженная жировая дистрофия приводит к декомпенсации сердечной деятельности. В почках липиды (нейтральные жиры, холестерин и фосфолипиды) откладываются в клетках эпителия канальцев. Холестерин накапливается и в эпителии канальцев, и в строме. Выраженное нарушение обмена заканчивается гибелью клеток, и функция почки резко нарушается.
Гиноидная липодистрофия не относится к заболеваниям, но тоже связана с изменением жировой клетчатки и рельефа кожи. Встречается у 85–90% женщин и связана с особенностями строения жировой клетчатки и действием половых гормонов. Является вариантом нормы и развивается у женщин с ожирением при низкой физической активности. Гиноидная липодистрофия — невоспалительное изменение жировой клетчатки с локальной гипертрофией (увеличением) клеток жировой ткани. Часто эту форму в быту называют «целлюлит», но это неправильно.
Целлюлит — заболевание, острое воспаление мягких тканей, протекающее с отеком, покраснением и местной болезненностью. При неосложненном целлюлите поражается кожа и жировая клетчатка, при осложнениях, что встречается редко, воспаление распространяется и на фасции и мышцы.
Причины
Молекулярная генетика позволяет разделить врожденную и приобретенную формы липодистрофии. Все формы врожденной генерализованной жировой дистрофии являются наследственными и у 80% больных при обследовании обнаруживают мутации генов, которые кодируют разные звенья липогенеза — дифференцировку адипоцитов, формирование жировых капелек, синтез триглицеридов, рост и увеличение адипоцитов.
При отсутствии генетических мутаций предполагается аутоиммунный генез. Приобретенная генерализованная форма и приобретенная парциальная имеют аутоиммунное происхождение. Тем более, что у многих больных выявляются параллельно аутоиммунные заболевания и снижение уровня факторов комплимента С3 и С4, как и при аутоиммунных заболеваниях.
Если рассматривать болезнь Уиппла, то причиной заболевания является инфицирование Tropheryma whippelii, которое происходит фекально-оральным путем. Первичное инфицирование часто происходит в детстве и протекает без симптомов или в виде диареи. После продолжительного периода (десятилетия) нахождения Tropheryma whippelii в организме при определенных иммунологических изменениях в организме развивается местная хроническая форма или процесс генерализуется, проявляясь классическими симптомами.
Симптомы
Несмотря на разные формы липодистрофий и проявление заболевания в разном возрасте, клиническая картина схожа. Кроме потери жировой ткани, заболевание протекает с выраженной инсулинорезистентностью, диабетом, повышением уровня триглицеридов и жировой дистрофией печени.
На первый план выступают изменения внешности — отсутствие жировой клетчатки (липоатрофия) на конечностях, лице (худое лицо) и мускулистая фигура («спортивный» внешний вид). При этом жировая клетчатка перераспределяется и максимально локализуется в области плеч, шеи и живота, у некоторых больных отмечается избыточный вес. При врожденной генерализованной форме обращают внимание на:
- контурированные мышцы;
- выступающие вены под кожей;
- увеличенные стопы и кисти (акромегалия);
- высокорослость;
- увеличение нижней челюсти;
- курчавые волосы;
- подчеркнутые кости орбиты;
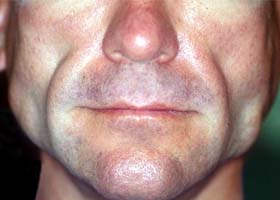
- черный акантоз кожных складок (повышенная пигментация кожи в паху и подмышечных впадинах);
- ксантомы на теле (бляшки желтоватого цвета из-за отложения липидов);
- гирсутизм (повышенное оволосение у женщин).
У всех больных выявляется увеличение печени, которое зависит от степени жировой дистрофии, повышение уровня триглицеридов. Резистентность к инсулину развивается в течение первых десяти лет, а сахарный диабет после пятнадцати лет.
В разные периоды жизни симптомы отличаются. В первый год жизни уже есть истончение жирового слоя и нарушения роста (либо отставание, либо гигантизм), задержка физического развития. В дальнейшем быстрый рост становится заметным и ребенка появляются акромегалоидные черты: крупные размеры нижней челюсти, стоп и кистей, повышенный аппетит, курчавые волосы, задержка интеллекта (хотя умственная отсталость не является непостоянным симптомом).

Фото детей с липодистрофией
После 10 лет формируется резистентность к инсулину, гиперпигментация складок. В возрасте 15 лет у многих развивается сахарный диабет, который проявляется потерей веса, жаждой, повышенным выделением мочи, кожным зудом. В пубертатном периоде выявляется крупный размер половых органов. В зрелом возрасте у женщин отмечаются нарушения цикла (аменорея, отсутствие овуляции), поликистоз яичников.
Болезнь Уиппла протекает в несколько стадий. На первой стадии повышается температура, увеличиваются лимфоузлы и появляются боли в суставах, то есть первая стадия протекает с общими симптомами. Во второй стадии (через 5-10 лет) наблюдаются кишечные нарушения: диарея, нарушение переваривания и всасывания. В связи с этим снижается вес и развивается дефицит витаминов. В третьей стадии развивается полисерозит — генерализованное воспаление серозных оболочек (плевры, перикарды, брюшины иногда со скоплением жидкости в полостях), что сопровождается характерными симптомами. Также поражается нервная система.
Наиболее характерными симптомами этого заболевания являются боли в животе, диарея, слабость, потеря веса. Стул обильный, бывает с примесью крови до 5–10 раз в сутки. Часто у больных появляется вздутие живота. Интенсивность боли в животе различная — чаще всего это распирание в животе после еды, а иногда — сильные спастические боли. Больной также отмечает, что у него постоянный кашель и длительное время болят суставы и мышцы. Чаще воспаляются голеностопные и коленные суставы, реже — локтевые, плечевые и лучезапястные. Может поражаться один или сразу несколько суставов при этом деформация их отсутствует. Боль в суставах беспокоит периодически в виде атак, которые сопровождаются повышением температуры. Суставной синдром ошибочно принимают за ревматизм.
Внекишечные проявления болезни Уиппла включают:
- Симптомы надпочечниковой недостаточности: низкое давление, отсутствие аппетита, пигментация кожи лица и шеи, тошнота и рвота.
- Кожные проявления в виде узелковых высыпаний.
- Поражение зрения: покраснение глаз, боли, рези, ухудшение зрения.
- Боли в сердце стенокардического характера, аритмии, одышка за счет миокардита и перикардита.
- Поражение нервной системы: потеря слуха, парез лицевого нерва, редко — утрата зрения, нарушение чувствительности кожи, парестезии.
Анализы и диагностика
- Осмотр больного выявляет характерные внешние признаки заболевания: отсутствие жировой клетчатки, «скелетированное» лицо, контурированные мышцы, выступающие подкожные вены, увеличенные стопы и кисти, черный акантоз естественных складок.
- Биохимический анализ крови. У всех больных рано выявляется повышенный уровень липидов, инсулина и сахара, глюкозотолерантный тест подтверждает снижение всасывания углеводов. Повышение уровня липидов за счет липопротеидов очень низкой плотности. При уровне глюкозы более 10 ммоль/л у больных повышается диурез и в моче появляется сахар.
- При развитии жирового гепатоза повышается уровень ферментов печени (АЛТ и АСТ).
- Исследование суточного выделения кортизола с мочой для выявления гиперкортицизма.
- Молекулярно-генетическое исследование. ДНК диагностика прямым секвенированием генов, связанных с заболеванием (BSCL2, AGPAT2, CAV1, PTRF), выявляет мутации, что важно для планирования потомства. Также проводится генетическая инвазивная пренатальная диагностика. Анализ проводится на панели липодистрофии.
- Электрокардиография выявляет аритмии различного типа и расширение комплекса QRS.
- Ультразвуковое исследование органов брюшной полости. Увеличение печени разной степени выраженности и диффузные изменения, соответствующие жировому гепатозу, увеличение селезенки, микролиты в почках (при мочекаменной болезни, которая встречается очень часто у этих больных).
- МР-спектроскопия печени — наличие жирового гепатоза.
- УЗИ малого таза. У женщин выявляют мультифолликулярные яичники и признаки хронического отсутствия овуляции.
- Эхокардиография обнаруживает кардиомегалию (увеличение размеров сердца).
При болезни Уиппла назначаются следующие обследования:
- Клинические анализы крови и мочи, биохимические анализы. Значительно повышены лейкоциты, тромбоциты СОЭ, снижен гемоглобин. Повышен уровень СРБ. Из-за нарушения мальабсорбции (нарушение усвоения веществ) снижается уровень в крови железа, калия, белка, кальция и холестерина.
- Колоноскопия. Определяется отек, покраснение и утолщение складок тонкой кишки. Неровность рельефа слизистой связана с отложением множественных желтоватоватых бляшек жиров.
- Биопсия кишки. При микроскопии биоптатов двенадцатиперстной и тощей кишок видны короткие ворсинки, наполненные лимфой. Также в слизистой оболочке отмечается внутри- и внеклеточное скопление жира и расширение лимфатических сосудов. В собственной пластинке оболочки много пенистых макрофагов (PAS-позитивные макрофаги с продуктами распада поглощенных бактерий). Такие же макрофаги находят в лимфоузлах брюшины, печени, синовиальной оболочке, селезенке, клапанах сердца, ткани мозга.
- ПЦР. Для этой реакции используют слизистую двенадцатиперстной кишки, биоптаты лимфоузлов, ликвор и синовиальную жидкость, полученную при пункции. Имеется вероятность получения ложноотрицательных и ложноположительных результатов.
- Компьютерная томография. Выявляет утолщение стенок кишки и увеличение лимфоузлов.
Лечение
Лечение липодистрофий
Специфического лечения не существует, поэтому проводится консервативное симптоматическое лечение, которое уменьшает проявления и предупреждает осложнения. Для этого больным назначаются:
- Диета с пониженным содержанием жиров и углеводов. Такая диета снижает нагрузку на печень и весь организм. Антиатерогенной диеты больные должны придерживаться постоянно, так как в большинстве случаев гиполипидемические средства неэффективны.
- Регулярные физические нагрузки.
- Лечение метаболических осложнений. Для симптоматического лечения назначают метформин (Глюкофаж), росиглитазон (Авандия), пиоглитазон (Диаб-норм, Пиоглит, Пиоглар) или вилдаглиптин (Галвус, Агарта, Вилдаглиптин-СЗ). При прогрессировании нарушений углеводного обмена применяется комбинация метформина с глитазонами (Актос, Диаб-норм, Пиоглар, Авандия).
- Для лечения дислипидемии — статины (Симвастатин, Аторвастатин, Ловастатин, Розувастатин, Флувастатин) и фибраты (Фенофибрат Канон, Липантил, Трайкор, Безамидин, Липо-мерц ретард).
- Плазмаферез. При недостаточной эффективности гиполипидемических средств 1 раз в месяц проводится плазмаферез. Курс состоит из 7-8 процедур. Данная процедура положительно влияет на липидный (снижается холестерин и триглицериды) и углеводный обмен (снижается уровень сахара в крови). После курса плазмафереза возобновляется прием препаратов фибратов и статинов. Через время процедуру плазмафереза повторяют 1 раз в месяц.
- При повышенном давлении проводится гипотензивная терапия.
- Гепатопротекторы при гепатомегалии при повышении уровня трансаминаз.
- Заместительная терапия адипокинами. При генерализованных наследственных липодистрофиях и у некоторых больных с семейной парциальной жировой дистрофией обмен веществ поддерживается препаратами лептина при низком его уровне в организме. Однако, при приобретенной парциальной форме генно-инженерный препарат неэффективен. Применяется Метрелептин, Миалепта — человеческий лептин.
- Заместительное лечение позволяет достичь компенсации липидного и углеводного обменов. Прием этого препарата снижает аппетит, уменьшает глюконеогенез в жировой ткани и печени, повышает усвоение глюкозы в мышцах, уменьшает отложение жиров в печени. Большинство пациентов, которым назначается метрелептин — дети и подростки.
При умственной отсталости ребенка показана работа с психологом и занятия по специальной программе.
При данном заболевании женщинам противопоказан прием оральных эстрогенов.
Лечение болезни Уппла
Основным методом лечения этого инфекционного заболевания является длительная антибактериальная терапия, которая проводится до двух лет. Обычно применяются тетрациклины, антибиотики пенициллинового ряда, бисептол и фторхинолоны. При отсутствии эффекта на антибактериальное лечение применяется гамма–интерферон.
Выраженная положительная динамика отмечается при ранней постановке диагноза и раннем начале лечения. Уже к концу 1–й недели устраняются лихорадка и диарея, суставной синдром — к концу 1-го месяца, также больной прибавляет в весе и его общее состояние улучшается. Неврологическая симптоматика устраняется медленнее. При неврологической симптоматике лечение начинают с внутривенного введения антибиотиков, проникающих в спинномозговую жидкость. При всех формах после активного лечения антибиотиками длительно назначается ко-тримоксазол (Бисептол, Двасептол, Бактрим). Этот комбинированный препарат содержит два активных противомикробных вещества (сульфаметоксазол и триметоприм) и хорошо предупреждает развитие неврологических осложнений.
Для устранения диареи и болей в животе назначают спазмолитики, имодиум, лоперамид, препараты висмута и энтеросорбенты. Для улучшения пищеварения показаны ферментные препараты: Паргрол, Фестал, Креон и пробиотики курсами.
С учетом синдрома мальабсорбции проводится коррекция водно–электролитного обмена (внутривенное капельное введение солевых растворов, глюкозы, альбумина) и устранение дефицита витаминов (внутримышечные инъекции витаминов В и С, питьевые ампулы глюконата железа — Тотема).
При генерализации процесса как сопутствующее лечение назначается Преднизолон (30–40 мг/в день со снижением дозы). При наличии ревматоидного артрита или анкилозирующего спондилита — цитостатики (Метотрексат, Арава, Имуран, Эндоксан) и моноклональные антитела (Ремикейд).
Доктора



Лекарства
 Глюкофаж
Глюкофаж Симвастатин
Симвастатин Аторвастатин
Аторвастатин Бисопролол
Бисопролол Эналаприл
Эналаприл Периндоприл
Периндоприл- Сахароснижающие препараты: Глюкофаж, Авандия, Диаб-норм, Пиоглит, Пиоглар, Галвус, Агарта, Вилдаглиптин-СЗ, Актос.
- Гиполипидемические средства: Симвастатин, Аторвастатин, Ловастатин, Розувастатин, Флувастатин, Фенофибрат Канон, Липантил, Трайкор, Безамидин, Липо-мерц ретард.
- Гипотензивные препараты: Бисопролол, Эналаприл, Периндоприл, Перинева, Престариум, Амлодипин + Периндоприл СЗ.
- Гепатопротекторы: Гептрал, Урсосан, Гептор, Адеметионин-Виал, Гептразин.
- Аналог гормона лептина: Миалепта.
Процедуры и операции
При жировой дистрофии могут понадобиться косметические операции:
- аутотрансплантация жировой ткани;
- применение наполнителей объема — филлеров поли-L-молочной кислоты, которые дают немедленный эффект восполнения объема;
- имплантат на основе гидроксиапатита кальция, косметический эффект от которого более стойкий (18–24 месяца).
У детей
Основные принципы симптоматического лечения сходны с таковыми у взрослых. Однако нужно отметить, что у детей наиболее перспективно и более часто применяется заместительное лечение метрелептином.
Лечение эффективно как при кратковременном, так и при долгосрочном применении, а происходящие изменения метаболизма значительно улучшают качество жизни больных и влияют на продолжительность жизни:
- уменьшаются микрососудистые осложнения диабета;
- половина больных, спустя один год лечения, прекращают использовать инсулин;
- снижается уровень триглицеридов;
- улучшается состояние паренхимы печени, что подтверждается гистологически;
- нормализуется процесс полового развития (не происходит преждевременное созревание).
Своевременное начало заместительного лечения защищает от прогрессирования метаболических расстройств до наступления пубертата.
Диета

Диета 10-й стол
- Эффективность: лечебный эффект через 1 месяц
- Сроки: постоянно
- Стоимость продуктов: 1700-1850 руб. в неделю

Диета при повышенном сахаре в крови
- Эффективность: лечебный эффект через 14 дней
- Сроки: постоянно
- Стоимость продуктов: 1400-1500 рублей в неделю

Гипохолестериновая диета
- Эффективность: лечебный эффект через 2 месяца
- Сроки: 3 месяца и более
- Стоимость продуктов: 1700-1900 руб. в неделю

Стандартная гиполипидемическая диета
- Эффективность: лечебный эффект через 3 месяца
- Сроки: постоянно
- Стоимость продуктов: 1800-1900 руб. в неделю
Диета при липодистрофиях — основа лечения метаболических осложнений. Больным приписывается гипокалорийная диета (1500 ккал/сутки), в которой строго ограничиваются животные жиры (сливочное масло, бараний, говяжий, свиной жир), легкоусвояемые углеводы (конфеты, сладкая выпечка, печенье и различные сладости). При этом содержание пищевых волокон (отруби, овощи, несладкие фрукты, цельнозерновые продукты) увеличивается. Больные могут придерживаться гипохолестериновой диеты (стандартная гиполипидемическая диета) или средиземноморской диеты.
Питание при болезни Уиппла должно обеспечивать поступление всех питательных веществ. Для коррекции мальабсорбции рацион больных должен содержать увеличенное количество животных белков (не менее 150 г), но ограничивать до 30 г жиры. При значительных метаболических нарушениях и потере веса иногда назначают даже смеси для энтерального питания или парентерального введения (альбумин/смеси аминокислот).
Профилактика
Пациентам с наследственными формами важно знать, что заболевание может быть у их детей. Для установления возможности рождения ребенка с нарушением жирового обмена членам семей, имеющим данную патологию, на этапе планирования беременности необходимо обследование у врача-генетика.
Для выявления врожденной патологии у ребенка проводится инвазивная пренатальная диагностика во время беременности. Материалом для ДНК диагностики служат ворсины хориона, полученные при биопсии или околоплодные воды, полученные при амниоцентезе (пункция амниотической оболочки). ДНК диагностика позволяет выявить характерный генный дефект.
Последствия и осложнения
- Липоатрофический диабет и его осложнения (микроангиопатия сетчатки, диабетическая полинейропатия нижних конечностей).
- Дислипидемия, которая осложняется заболеваниями печени — жировой гепатоз, стеатигепатит, цирроз, гепатоцелюллярная карцинома и портальной гипертензией уже в раннем возрасте.
- Панкреатит, связанный с дислипидемией.
- Нарушения менструального цикла (аменорея) и репродуктивной функции. У женщин в ряде случаев поражаются яичники (поликистоз), это ведет к бесплодию. Вторичный гипогонадизм у мужчин.
- Сердечно-сосудистые заболевания (кардиомиопатия).
- Патология почек.
Основными причинами смерти при липодистрофии являются:
- Сердечная недостаточность на фоне кардиомиопатии и аритмий.
- Инфаркт миокарда.
- Печеночная недостаточность.
- Почечная недостаточность.
- Желудочно-кишечные кровотечения.
- Острый панкреатит.
Прогноз
Прогноз при липодистрофиях неопределенный и зависит от вовлечения в процесс печени, сердца, и почек. В тяжелых случаях развивается цирроз печени, сердечная недостаточность и почечная недостаточность, при которых прогноз неблагоприятный. Основными инвалидизирующими факторами, а также причинами смерти являются осложнения: сахарный диабет, острый панкреатит и цирроз, как исход длительно протекающего стеатоза печени. При невыраженных нарушениях функции печени, почек и сердца прогноз относительно благоприятный.
Прогноз при болезни Уиппла при раннем выявлении и правильном лечении благоприятный. Рецидивы возникают в 10–35% случаев, при которых проводится лечение, как и при первичном проявлении заболевания. Вероятность рецидивов есть даже в случае эрадикации возбудителя при первом эпизоде болезни. В связи с этим многими врачами предлагается проведение пожизненной профилактики доксициклином. Если изначально лечение (длительная антибактериальная терапия) не проводится, то пациенты умирают через 1,5–2 года со времени развития кишечных симптомов. Смерть наступает в связи с поражением миокарда или от надпочечниковой недостаточности. Неблагоприятный прогноз при неврологических осложнениях, которые возникают при рецидивах или при первично нераспознанном и нелеченом заболевании.
Список источников
- Соркина Е.Л., Тюльпаков А.Н. Наследственные и приобретенные липодистрофии: молекулярно-генетические и аутоиммунные механизмы. Ожирение и метаболизм. 2018; 15(1): 39-42.
- Липодистрофия тотальная врожденная. Клинические рекомендации. Российское общество медицинских генетиков 2017, 18 с.
- Соркина Е.Л., Калашникова М.Ф., Мельниченко Г.А., Тюльпаков А.Н. Семейная парциальная липодистрофия (синдром Dunnigan) вследствие мутации в гене LMNA: первое описание клини ческого случая в России. Терапевтический архив. 2015; 87(3):83-87.
- Скворцов В. В., Тумаренко А. В., Павлов В. К. Диагностика и лечение болезни Уиппла/ Экспериментальная и клиническая гастроэнтерология. 2018; 153 (5): 123–127.
- Белов Б. С. Болезнь Уиппла / Современная ревматология 2013, №1, с. 12–17.
Комментарии



Похожие болезни









Последние комментарии
Вероника: Все понятно,но действительно что при ПН можно например рис,компот,мед при диабете ...
Елизавета: Несколько месяцев назад, спустя почти 2 года свободы, у меня появились отношения. Вместе ...
Людмила: Гусиная кожа это не заболевание, а состояние кожи, и улучшить его можно увлажнением ...
вера: Сыну было 4 мес, путал день с ночью, укачивала часами, решила попробовать Дормикинд ...