Синдром Шерешевского-Тернера

Общие сведения
Синдром Шерешевского-Тернера — это хромосомное геномное заболевание. Какая мутация связана с этим заболеванием? Данный синдром характеризуется полным или частичным отсутствием второй половой X-хромосомы. В 60% случаев у больных отсутствует одна X-хромосома (полная моносомия), а в остальных случаях отмечаются мозаичные варианты (хромосомный набор частично сохранен) или структурная аномалия этой хромосомы (делеции, транслокации и другие). Какой пол подвержен этой патологии? Две Х хромосомы связаны с женским полом, поэтому заболевание проявляется у девочек — 1 случай на 2500 новорожденных. Мужской пол имеет ХY хромосомы и вероятность заболевания у мальчиков низкая. Заболевание не наследственное, но, если в роду имеются случаи хромосомных заболеваний, риск заболевания детей повышается.
Опасность этого заболевания состоит в том, что эмбрионы с кариотипом 45X0 (моносомия Х) очень редко достигают стадии плода (9-й недели) — происходят спонтанные аборты. Родившиеся дети имеют множественные врожденные аномалии, самыми опасными из которых являются врожденные пороки сердца, нередко становящиеся причиной ранней смерти больных. У девочек задерживается или полностью отсутствует половое созревание, в результате чего они бесплодны. При кариотипе 45,Х/ 46,ХY развивается гонадобластома.
Патогенез
У человека половая клетка имеет 23 пары хромосом (всего 46), в которых локализованы гены. Появление синдрома Шерешевского-Тернера связано с тем, что половая клетка имеет неправильный набор хромосом — кариотип человека состоит из 45 хромосом. Под влиянием различных факторов нарушается гаметогенез и после оплодотворения зигота имеет неправильное количество хромосом. Мутация синдрома Шерешевского-Тернера связана именно с Х хромосомой — она только одна (45,X, моносомия), или их две, но одна измененная (45,X/46,XX мозаицизм).
Эмбрионы с моносомией в 99% погибают на разных стадиях, чаще всего к 6 неделям. Только 1% плодов с моносомией выживает и у них внутриутробно обнаруживают признаки заболевания: дефекты нервной трубки, задержка развития, отек шеи, легких и общий отек плода. Нередко встречается порок коартация аорты. Лимфатические отеки появляются на фоне аномалии лимфатических сосудов — аплазия сосудов и гипоплазия анастомозов, которые соединяют глубокие и поверхностные лимфатические сосуды. Лимфатический отек обусловливает появление складок задней поверхности шеи и изменения аорты. Гормональные нарушения связаны с гипоплазией яичников и недостаточной выработкой половых гормонов. Недостаток эстрогенов у подростков не активизирует гормон роста и инсулиноподобный фактор роста 1, поэтому костеобразование идет недостаточно и формируется остеопороз.
Классификация
Варианты хромосомной аномалии:
- Моносомия X (45,XО). Отсутствует одна Х-хромосома во всех клетках, что объясняется сбоем во время их деления. Потеря материнской Х-хромосомы проявляется более тяжелыми пороками внутренних органов. Чаще отсутствует отцовская Х-хромосома. При кариотипе 45,ХО нарушается рост и отмечается гипогонадизм в 100% случаев. Замедление роста видно уже на 1-м году жизни, а заметное отставание — после 4-х лет. У подростков с кариотипом 45,ХО молочные железы отсутствуют, лобковое оволосение развивается к 12-13 годам, но оно скудное, отмечается первичная аменорея. У взрослых женщин — гипоплазия матки и дизгенезия гонад.
- Структурные аномалии (делеция короткого или длинного плеча, изохромосома и кольцевая хромосома). Делеции одной из хромосом вызывают невыраженные аномалии по сравнению с полной моносомией. У таких женщин отмечается дисгенезия гонад, низкий рост. Делеции длинного плеча влияет на половое развитие, но серьезные нарушения внутренних органов отсутствуют.
- Мозаичный кариотип 45,XО/46,XX. Хромосомный набор сохранен частично: одни клетки имеют две Х- хромосомы, а другие — только одну или измененную. Это наиболее благоприятный вариант, поскольку рост у больных страдает незначительно, отмечается частичный пубертат и в некоторых случаях возможна беременность. Также возможен мозаицизм, связанный с У-хромосомой — 45,ХО/46,ХУ. При этом варианте отмечается мужской гермафродитизм — наружные гениталии формируются по женскому фенотипу, но с измененным клитором, яички недоразвиты. В данном случае нет возможности развития мужского пола, поэтому терапевтические и хирургические вмешательства направлены на приближение к женскому полу.
Из всех вариантов доминируют чистая моносомия и мозаичный вариант, реже встречаются варианты структурных изменений.
С количественным изменением Х хромосомы (в сторону их увеличения) связаны синдром Клайнфельтера и синдром Дауна. Синдром Клайнфельтера — полисомия по Х-хромосоме: кариотип 47 XXY является классическим и встречается чаще, а кариотипы 48 XXXY, 49 XXXXY и 48 XXYY — реже. Присутствие в кариотипе Y-хромосомы определяет мужские половые органы, но лишние Х-хромосомы сказываются на повреждении яичек, понижении продукции андрогенов и бесплодии у мужчин. Изменения проявляются в пубертатном возрасте: высокорослость, длинные конечности, гинекомастия, евнухоидное телосложение. Евнухоидное телосложение (высокая талия, широкий таз, длинные ноги, ожирение по женскому типу, скудное оволосение) характерно для классического варианта синдрома.
Синдром Дауна — трисомия 21 хромосомы, которая проявляется многочисленными аномалиями: плоское лицо, короткий нос, монголоидные глаза, плоская переносица, плоский затылок, эпикант, брахицефалия, открытый рот, широкая шея, высокое небо, аномалии зубов. Характерны короткие конечности, повышенная подвижность суставов, укорочение пальцев, искривление V пальца. После 8 лет у больных часто появляется катаракта.
Причины синдрома Шерешевского-Тернера
Основные причины появления заболевания — количественная или структурная аномалия Х-хромосомы. Что влияет на это, неизвестно. Не подтверждено и влияние на этот процесс возраста родителей. X-хромосомный мозаицизм связан с нарушением дробления зиготы. Та или иная генная мутация отражается на количестве аномалий и течении заболевания. На коротком плече располагаются гены, при делеции которых развиваются почти все внешние признаки данного синдрома. В Х-хромосоме локализуются несколько генов, которые связаны с нормальным развитием и функцией яичников.
Кольцевая Х-хромосома образуется при утрате концов длинного и короткого плеча и замыкании их в кольцо. Эта аберрация встречается в мозаичной форме и проявляется у некоторых больных аномалиями лица, сращением пальцев, тяжелой умственной отсталостью, а классические признаки синдрома отсутствуют. У остальных больных выявляют характерные признаки рассматриваемого синдрома.
Симптомы
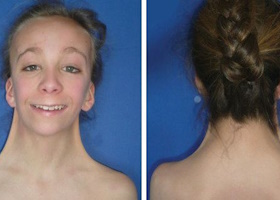
Характерные признаки — различные аномалии развития: низкий рост, короткая шея, широкая грудная клетка, низкий рост волос сзади, низко посаженные и большие уши, маленькая челюсть и аномалия зубов, высокое небо, птоз (опущение века) и эпикантус («монгольская складка» глаз), коарктация аорты (сужение просвета аорты). Также характерны аменорея или нерегулярные менструации, бесплодие, гипотиреоз, повышенная подвижность суставов, инсулинорезистентность. У взрослых выявляются искривление коленных и локтевых суставов и остеопороз.
Дети рождаются маловесными и длиной тела меньше, чем у других новорожденных. Уже при рождении выявляется лимфатический отек кистей и крыловидные складки на шее (избыток кожи).

Нарушение роста наблюдается в 97-100% случаев. Отставание отмечается уже в утробе задержкой роста по сравнению с нормально развивающимися плодами. С 3 лет становится заметной малая прибавка роста, а в пубертатном возрасте отсутствует ростовой скачок. В конечном счете к окончанию периода роста женщина имеет рост 140-145 см, что на 20 см ниже, чем у здоровых.
Недостаточность яичников приводит к тому, что не увеличиваются молочные железы, матка и влагалище. Но под влиянием андрогенов надпочечников к 12-13 годам развивается оволосение. После 12 лет созревание скелета задерживается, что связано с недостатком эстрогенов.
Со стороны кожи — множественные доброкачественные невусы, а со стороны волос — гипертрихоз (усиленный их рост на теле). Невусы появляются после 4 лет и размеры их различны — от практически незаметных до крупных. В период пубертата невусы начинают расти. Иногда встречается гипертрихоз, выраженный на предплечьях. Также возможно выпадение волос на голове, витилиго на разных участках тела. Ногти у больных узкие, короткие и сидят глубоко на ногтевом ложе.
Лимфатический отек стоп и кистей появляется у новорожденного в родильном доме. Он держится несколько дней, но может быть до 3 лет. В зрелом возрасте отеки исчезают, но при нагрузке (при длительной ходьбе или стоянии) снова появляются. Со стороны костного скелета характерным является укорочение IV и V пальцев рук и III—V пальцев стоп, искривление V пальца кистей. Отмечается искривления костей голени, локтевых и коленных суставов.
Основным признаком коарктации аорты является повышение давления на руках и невозможность его определить на ногах. Дефект межпредсердной перегородки проявляется одышкой при нагрузке, сердцебиением и аритмией, что связано с перегрузкой правой половины сердца и сосудов легких. Пороки мочевыделительной системы встречаются у 40% больных: подковообразная почка, аномалии расположения, гипоплазия почек с двух сторон, пороки чашечек и лоханки, удвоение лоханки и мочеточников. У женщин при этом заболевании отмечается ожирение, нарушение толерантности к углеводам и сахарный диабет. Нарушение обмена холестерина влечет риск развития атеросклероза и ишемической болезни сердца. Со стороны нервной системы — снижение слуха, остроты зрения, эмоциональная лабильность, олигофрения.
Анализы и диагностика
- Диагноз устанавливается при исследовании кариотипа. Нормальная кариограмма человека — во всех клетках 46, XY (мужской пол) или 46, ХХ (женский пол). Изучение кариотипа плода назначается при обнаружении следующих УЗИ-признаков: утолщение затылочного изгиба коарктация аорты, брахицефалия, аномалии строения почек, задержка развития плода, аномалии строения левых отделов сердца, водянка мозга. Возможный кариотип при данном заболевании: мозаицизм (45,XО/46,XX и редко 45,ХО/46,ХУ) или моносомия (45,ХО).
- После рождения при установленном диагнозе важно следить за развитием возможной патологии и в зависимости от возраста назначаются: от 0 до 4 лет — ЭхоКГ, осмотр тазобедренных суставов для выявления дисплазии, от 4-10 лет — гормоны щитовидной железы, скрининг на целиакию, после 7 лет показано обследование у ортодонта; детям старше 10 лет — тиреоидный статус, печеночные маркеры, уровень глюкозы, липидный спектр, креатинин, мочевина, половые гормоны для начала заместительной терапии, определение ИФР-1 при лечении гормоном роста, рентгенография кисти и запястья для определения костного возраста и выявления остеопороза. После 15-16 лет определение минеральной плотности кости, УЗИ матки и яичников, коагулограмма при назначении половых гормонов.
Взрослым женщинам проводят следующие обследования:
- Определение уровня фолликулостимулирующего (маркер функции яичников), лютеинизирующего гормона и эстрогенов. Отмечается повышенный уровень гонадотропинов (ФСГ и ЛГ) и снижение эстрогенов.
- УЗИ яичников и матки. Гонады взрослых женщин выглядят соединительнотканными тяжами или рудиментами гонад яйцеклеток, матка маленьких размеров.
- Магнитно-резонансная томография малого таза. Регистрирует отсутствие фолликул в яичниках и функциональные кисты яичников в виде жидкостных камер.
- ЭхоКГ и ЭКГ для выявления пороков развития.
- Гормоны щитовидной железы и тиреотропный гормон.
Лечение
Специфическое лечение синдрома отсутствует, а симптоматическая терапия включает несколько этапов:
- назначение анаболических стероидов и инъекций препаратов гормона роста для увеличения роста;
- эстрогенсодержащие препараты с возраста полового созревания для появления вторичных половых признаков;
- профилактика и лечение остеопороза;
- оперативное лечение пороков;
- коррекция питания.
Лечение низкорослости
В детском и подростковом возрасте заболевание синдром Шерешевского-Тернера нуждается в коррекции низкорослости. До 12-13 лет назначаются анаболические стероиды (нерабол, ретаболил) и инъекции препаратов рекомбинантного гормона роста. Последние назначаются по схеме: ежедневно подкожно, перед сном в дозе 0,05 мг/кг. Чем длительнее проводится это лечение, тем больше конечный рост. Лечение гормоном роста заканчивают, когда костный возраст соответствует 15 годам. Гормон роста применяют самостоятельно или в сочетании с эстрогенами и такая комбинация более эффективно отражается на росте.
Заместительная терапия половыми гормонами
Ее можно начинать по достижению костного возраста 12 лет. Назначение эстрогенов в минимальных дозах положительно влияют на развитие половых органов, молочной железы, обеспечивает пубертатный скачок роста и является профилактикой остеопороза. На фоне гормонального лечения улучшается и психическое состояние.
При лечении гормоном роста и эстрогенотерапии достоверно увеличивается конечный рост. При проведении такого лечения девочка должна обследоваться у эндокринолога и гинеколога каждые полгода. Кроме эстрогенов таблетированных используются трансдермальные эстрогены или депо-формы. Дозу эстрогенов в течение 2-3 лет повышают, после этого срока или с появлением менструации к лечению добавляют прогестин. Синтетические оральные контрацептивы в подростковом возрасте не рекомендуется использовать, поскольку содержание синтетических эстрогенов в них высокое, а синтетические прогестины препятствуют развитию половых органов.
У взрослых женщин для регуляции менструального цикла применяются комбинированные контрацептивы, содержащие дезогестрел + этинилэстрадиол (Мануэль, Мерсилон, ПаниЖенс дезо), на фоне которых при мозаичном варианте цикл нормализуется. Заместительное лечение эстрогенами молодым женщинам проводится постоянно и преимущество в этом имеют трансдермальные препараты в виде геля (эстрадиолсодержащий гель — Дивигель).
Лечение бесплодия
Назначается Кломифен (стимулятор продукции гонадотропинов) одновременно с хорионическим гонадотропином в инъекциях (стимулирует образование эстрогенов и прогестерона, вызывает овуляцию).
Доктора



Лекарства
 Растан
Растан Джинтропин
Джинтропин Дивигель
Дивигель Мерсилон
Мерсилон- Препараты рекомбинантного гормона роста: Омнитроп, Растан, Джинтропин, Нордитропин, НордиЛет, Генотропин, Хуматроп, Сайзен.
Процедуры и операции
Врожденные пороки и эстетические недостатки нуждаются в коррекции. При коарктации, как наиболее распространенном пороке при данном синдроме, выполняются два вида операций: резекция аорты (суженного участка) и сшивание аорты «конец в конец», по другому методу тоже производится резекция, но для восстановления целостности аорты применяется протез — синтетический трансплантат.
При лимфатических отеках иногда прибегают к хирургическому вмешательству — выполняют ангиопластику, хотя эффективность ее не доказана. Удаление невусов имеет косметическое значение, но всегда учитывают риск развития у этих больных келоидных рубцов. В связи с этим многие отказываются от косметической коррекции. К коррекции аномалий прибегают при значительном косметическом дефекте — крыловидные складки на шее или выраженный птоз, который затрудняет функции глаза.
Диета
Питание больных должно быть полноценным по содержанию белков, жиров, углеводов, микроэлементов и витаминов. Полноценный белок (яйца, мясо, рыба, творог, сыры) является основным строительным материалом для мышц и необходим в период активного лечения анаболическими стероидами и гормоном роста.
Коррекцию питания проводят, учитывая возможные осложнения заболевания. С учетом повышенного риска остеопороза и переломов питание в любом возрасте должно быть включать продукты, богатые кальцием (молочная продукция, орехи, кунжутное семя) и витамином Д (рыба, морепродукты, печень трески, рыбий жир). При избыточном весе, который отмечается у всех девушек и женщин при этом заболевании, и нарушении углеводного обмена показано питание с ограничением простых углеводов — сахар, мед, варенье, мороженое, кондитерские изделия, выпечка.
Профилактика
Профилактика этой патологии не разработана — предотвратить генетическую аномалию невозможно. Однако имеется возможность провести дородовую диагностику у плода. У женщин с мозаичным кариотипом возможна самостоятельная беременность за счет сохранившихся фолликулов и имеется риск рождения больного ребенка. Поэтому проводится неинвазивная пренатальная диагностика, устанавливающая риск заболевания плода, которая возможна после 10 недели беременности. Проводят забор крови беременной или соскоб со слизистой ее щеки. Диагностируют 112 наследственных заболеваний, включая Шерешевского-Тернера. Если у плода при УЗИ выявляют признаки синдрома, проводится инвазивная пренатальная диагностика (амниоцентез — забор околоплодных вод путем пункции или забор ворсин хориона), которая точно устанавливает хромосомный набор плода. Это дает возможность выявить патологию на ранних этапах и родители могут принять решение о продлении беременности или ее прерывании.
Последствия и осложнения
- Риск развития сахарного диабета и гипотиреоза.
- Яичниковая недостаточность и бесплодие. Это серьезная проблема при данной патологии. Менархе возникает самостоятельно в редких случаях, еще реже бывает спонтанная беременность, поэтому все пациентки нуждаются гинекологическом обследовании и лечении.
- Остеопороз и остеопения. Остеопения развивается в период пубертата и сохраняется во взрослом возрасте. У взрослых отмечается повышенный внутрикостный обмен и частота переломов выше, чем у здоровых. В большинстве случаев переломы встречаются в характерных для остеопороза местах — запястье, шейка бедра или позвоночник.
- Сердечно-сосудистая патология. Основная причина смерти — разрыв аорты выше места сужения или ее расслоение. Выявление коарктации аорты и проведение операции предупреждают эти грозные осложнения и смерть пациентки. Также больные находятся в группе риска по развитию артериальной гипертензии.
- Задержка психического развития, проблемы с интеллектом, вниманием и памятью.
- Развитие гонадобластомы (при кариотипе 45 Х/46 ХY).
Прогноз
При отсутствии тяжелых пороков прогноз для жизни благоприятный. В большинстве случаев люди с синдромом Шерешевского-Тернера при своевременном симптоматическом лечении ведут полноценную жизнь. Их состояние зависит от сочетания аномалии сердечно-сосудистой системы с гипотиреозом или сахарным диабетом, что в целом влияет не только на качество жизни, но и на ее продолжительность. Прогностически неблагоприятными являются врожденные пороки сердца, которые становятся причиной смерти.
Спонтанная беременность наступает редко и только у женщин с мозаичным кариотипом. В связи с развитием репродуктивных технологий у бесплодных женщин появилась возможность стать матерями при использовании донорских яйцеклеток и яйцеклеток самой женщины. Вынашивание и рождение ребенка возможно только на фоне гормональной терапии.
Список источников
- Волеводз Н.Н. Федеральные клинические рекомендации «Cиндром Шерешевского-Тернера (СШТ): клиника, диагностика, лечение». Проблемы Эндокринологии. 2014;60(4):65—76.
- Адамян Л.В., Сибирская Е.В., Кириллова Ю.А., Мурватова К.К., Серегина В.Ю. Репродуктивные исходы у пациенток с синдромом Шерешевского—Тернера (клинический случай). Проблемы репродукции. 2023;29(5):69—72.
- Черемискин В.П., Филянина А.В. Синдром Шерешевского-Тернера и беременность. Пермский медицинский журнал, 2021, Том38, №5, С. 70—78.
- Панкратова М. С., Петеркова В. А. Синдром Шерешевского-Тернера: особенности мониторинга в разные возрастные периоды. Доктор. Ру, 2009, №6(50), С. 46—51.
- Вяткина С. В., Кузнецова Т. В. Современные представления о синдроме Шерешевского-Тернера. Журнал акушерства и женских болезней, 2007, Том16, выпуск 1, С. 56—64.

Комментарии



Похожие болезни



Статьи по теме






Последние комментарии
Инна: Что-то меня дернуло и решила я попробовать новый гель для интимной гигиены. Баночка такая ...
Вероника: Все понятно,но действительно что при ПН можно например рис,компот,мед при диабете ...
Елизавета: Несколько месяцев назад, спустя почти 2 года свободы, у меня появились отношения. Вместе ...
Людмила: Гусиная кожа это не заболевание, а состояние кожи, и улучшить его можно увлажнением ...