Синдром Лея (болезнь Лея)


- Лечащие врачи: Невролог, Генетик, Репродуктолог (ЭКО)
- Диеты при болезни: Диета для нервной системы
Общие сведения
Синдром Лея относится к редким нейрометаболическим синдромам, поражающим ЦНС, и вызывающим нарушение координации движений и мышления и летальный исход. Заболевание наследственное по аутосомно-рецессивному типу или Х-сцепленному. Патологии чаще всего подвержены маленькие детки до 2-х лет – 1-2 ребенка на десятки тысяч, в более редких случаях – подростки и взрослые особы. Код по МКБ-10: G 31.8
Миру стало известно об этом заболевании благодаря Денису Лею, который описал его еще в 1951 году. В научных кругах обособить синдром от подобных энцефалопатий удалось в 1954 г. Связь с митохондриальной активностью смогли выявить 1968 г, но главным продвижением стало обнаружение мутации в цитохромоксидазах в 1977 г.
Проблема состоит в том, что эффективных препаратов или методов лечения болезни Лея до сих пор не разработано, ведь причин её множество, начиная с влияния мутаций в генах, отвечающих за работу митохондрий, и заканчивая – осложнениями дегенеративных заболеваний нервной системы, аномальными образованиями некротизированных очагов или прорастания сосудов, глиоза в структурах головного мозга.
Единственным профилактическим методом на сегодня является «рождение детей от трех родителей», то есть благодаря использованию донорских яйцеклеток и митохондриальной донации.
Патогенез
Для таких митохондриальных заболеваний как синдром Лея характерно полиорганное поражение с вовлечением нервных и мышечных тканей. В основе патологии обычно лежит сбой регулирования обмена пировиноградной кислоты, при этом наблюдается нарушение обменных реакций, снижение выработки энергии и замедление процессов перемещения электронов в клетках дыхательной системы. Процессы могут спорадически усилиться, чему способствует наследственные факторы. Для запуска развития синдрома необходимо присутствие в организме более 90% мутантной мтДНК от всей мтДНК. В случае меньшего содержания мутантной ДНК симптоматика напоминает нейропатию и сводится к атаксии и пигментному ретиниту.
Для болезни характерно раннее развитие и стремительное злокачественное течение с присоединением большого количества различных неврологических нарушений, затем начинаются проблемы с дыханием и метаболизмом.
Классификация
В зависимости от патогенеза и возрастных особенностей выделяют различные варианты нарушений, выражающиеся в виде:
- недостаточности цитохром С-кислород-оксидоредуктазы;
- некротизирующей подострой инфантильной энцефалопатии Лея;
- синдрома Лея у взрослых особ;
- синдрома NARP или дефекта митохондриальной ДНК;
- Х-сцепленного синдрома Лея;
- недостаточности фермента – пируваткарбоксилазы.
В зависимости от течения синдром Лея бывает:
- острой формы – в очень редких случаях и напоминает острую энцефалопатию;
- хроническая и подострая болезнь Лея.
Причины
Главным провоцирующим фактором синдрома Лея принято считать недостаточность цитохромоксидазы и мутации генов, отвечающих за работу митохондрий:
- замену Т (тимина) на Г (гуанин) или Ц (цитозин) в 8933-м положении 6-й субъединицы АТФ-синтазы;
- мутации в флавопротеиновой единице сукцинатдегидрогеназного комплекса, в SURF-генах либо в двух ядерных генах (приводит к развитию митохондриальных болезней второго класса), в целом – их описано около 40.
Однако, способствовать нейрометаболической энцефалопатии может:
- образование некротизированных очагов или онкоструктур (злокачественных либо доброкачественных) в структурах мозжечка или мозгового ствола;
- прорастания сосудов или развитие глиоза;
- наследственная предрасположенность и мутагенные факторы любой природы – биологические, экологические, химические и пр.;
- осложнение дегенеративных заболеваний, поражающих нервную систему;
- а также результате вакцинации.
Симптомы
Синдром Лея и его прогрессирование приводит:
- к возникновению тошноты и рвоты;
- к быстрому снижению массы тела и слабости;
- к развитию задержки психомоторных функций (например, снижению внимательности) и нарушениям сознания;
- к приступам тонико-клонических судорог либо мышечной дистонии/гипотонии;
- к возникновению респираторных аномалий;
- к атрофии зрительных нервов, возможно даже слепоте;
- к тугоухости;
- к повышению утомляемости и сонливости;
- а также к нарушениям координации, сухожильных рефлексов, актов глотания, тремору конечностей.
Клиническая характеристика синдрома Лея при других формах патологии
Клиническая характеристика и патогенез схожи, но симптоматика может дополняться:
- метаболическим компенсаторным ацидозом или приступами тяжелого ацидоза;
- снижением уровня бикарбонатов в крови;
- увеличением объемов молочной, пировиноградной кислоты в крови и ликворной жидкости;
- приступами миоклонии;
- гиповинтиляцией и развитием тахиапноэ;
- атаксией и клоническими подергиваниями;
- спастическими парезами;
- эпилептическими припадками;
- прогрессирующей деменцией;
- центральной скотомой;
- кардиомиопатией;
- симптомами экстрапирамидной и пирамидной недостаточности.
Анализы и диагностика
Болезнь Лея относится к подострым либо хроническим некротизирующим энцефаломиопатиям и является важным направлением изучения в детской невралгии. Для её диагностики необходима консультация у врача-невролога, а также:
- проведение общего анализа крови и её биохимических исследований, выявляющих лактатацидоз (повышенную концентрацию лактата, пирувата в кровяном русле и ликворе), снижение количества карнитина, активности цитохромоксидазы в культуральных клетках — фибробластах;
- изучение структур головного мозга посредством магнитно-резонансной томографии;
- изучение электрической активности (ЭЭГ) и морфологических структур головного мозга.
Лечение
Медикаментозное консервативное лечение болезни Лея обычно симптоматическое и включает использование:
- антибиотиков, например, Ампициллина;
- различных методов диализа;
- ноотропов;
- противосудорожных средств;
- препаратов, содержащих В1 и биотин.
Доктора



Лекарства
 Кавинтон
Кавинтон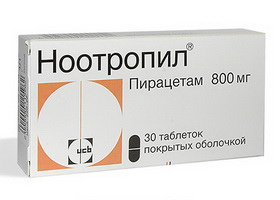 Ноотропил
Ноотропил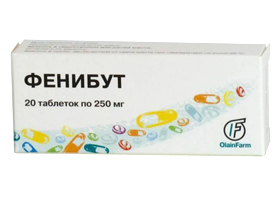 Фенибут
Фенибут Карбамазепин
Карбамазепин Ампициллин
Ампициллин- Кавинтон — антигипоксическое, нейропротективное, антиагрегационное, сосудорасширяющее средство, помогающее улучшить мозговое кровообращение. Стандартная суточная доза составляет 15-30 мг и должна быть разделена на три приема.
- Ноотропил — таблетки с ноотропным действием, позволяющие улучшить концентрацию внимания и когнитивные функции. Максимальная суточная доза 24 г, разделенная на 2-3 приема. С осторожностью следует принимать особам, имеющим проблемы с работой почек.
- Фенибут – еще один ноотроп, который выпускается в таблетках и помогает при различных психопатологических и соматовегетативных расстройствах, при головокружениях, бессоннице и пр. Детям достаточно давать полтаблетки три раза вдень.
- Карбамазепин – психотропное противоэпилептическое средство. Таблетки отпускаются только по рецепту, поэтому необходимо назначение врача и индивидуальный расчет дозы.
- Ампициллин – антибактериальный препарат из группы пенициллинов, помогает при различных инфекциях дыхательных и мочевыводящих путей, ЖКТ, менингите, эндокардите, сепсисе, коклюше и пр. Для детей суточную дозу высчитывают на массу тела — 100 мг/кг и назначают в 4–6 приемов.
Процедуры и операции
В результате нейродегенеративных процессов больным может понадобиться искусственная вентиляция легких, санация дыхательных путей, гастростомы, трахеостомы, кардиостимулятор и пр.
Самый эффективный метод для предупреждения появления ребенка с синдромом Лея – рождение от «трех родителей», которое подразумевает реконструкцию зиготы путем переноса ядер родителей в донорскую энуклеированную яйцеклетку.
Диета при синдроме Лея

Диета для нервной системы
- Эффективность: лечебный эффект через 2 месяца
- Сроки: постоянно
- Стоимость продуктов: 1700-1800 рублей в неделю
В первую очередь предполагает контроль количества поступающего в организм белка за день. Оно не должно превышать 1-1,5 г в сутки. Конечно же питание должно быть здоровым и рациональным. Если нарушены акты глотания, то пациентам может быть рекомендовано питание искусственным смесями через зонд. При этом необходим контроль и нормализация уровня витаминного и жирового обмена, недопущение развития ожирения. Больным следует избегать тяжелых физических нагрузок и постов, что может повысить нагрузку на митохондрии.
Прогноз
Для подострого и хронического течения характерен летальный исход в течение нескольких лет. Если заболевание развивается стремительно, то смерть чаще всего наступает в результате паралича дыхательного центра, находящегося в продолговатом мозге.

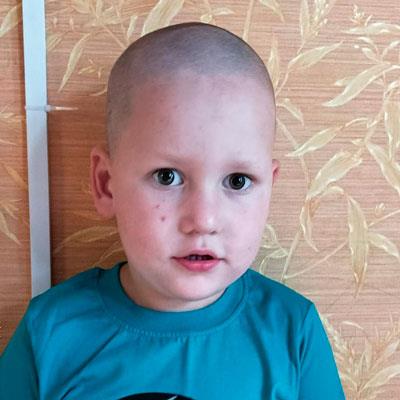
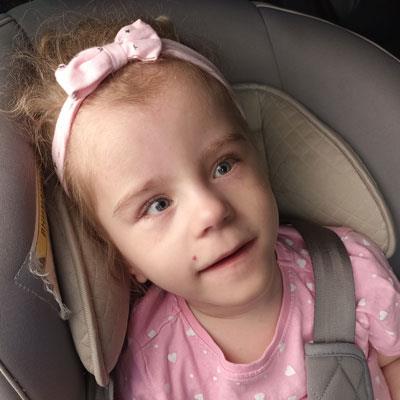
Семья Лейк с детьми, страдающими от болезни Лея
Пример — семья Лейк (на фото родители с дочкой Жасмин – слева и сыном Джейдом — справа), у которых родилось четверо детей с синдромом Лея, трое из которых уже умерли в малолетнем возрасте. Их дети так и не смогли научиться самостоятельно ходить и разговаривать, страдали от приступов и умственной отсталости, причиной тому был смертельный ген у их матери Адель, поэтому при планировании семьи крайне важно проходить ДНК-тестирование родителей и медико-генетическое консультирование у квалифицированных специалистов. Главное помнить, что синдром Лея и другие генетические заболевания намного легче предотвратить, нежели вылечить.
Список источников
- Гинтер Е.К. Медицинская генетика. М.: медицина,2003. -130 с.
- Николаева Е.А., Яблонская М.И., Барсукова П.Г. и др. Болезнь Лея, обусловленная мутацией гена SURFI: диагностика и подходы к терапевтической коррекции. Российский вестник перинатологии и педиатрии, 2006, №2, С. 27-31.

Комментарии





Похожие болезни



Статьи по теме




Последние комментарии
вера: Сыну было 4 мес, путал день с ночью, укачивала часами, решила попробовать Дормикинд ...
Сергей: Здраствуйте! Подскажите где можно достать этот препарат Денебол гель?
Надежда: Наконец нашла эту статью, где рассказано как правильно питаться, после удаления желудка ...
Алла Анатольевна: Как только прочитаешь противопоказания, сразу сдохнуть хочется.