Лейкодистрофия

Общие сведения
Лейкодистрофия головного мозга — врожденное заболевание нервной системы. Википедия дает следующее определение данной патологии: «это группа тяжелых наследственных заболеваний, которые характеризуются поражением белого вещества головного мозга спинного». Это нейродегенеративные, достаточно редкие заболевания, для которых характерно нарушение обмена веществ, связанного с врожденным дефектом того или иного фермента, что сопровождается накоплением метаболитов, которые вызывают разрушение миелина белого вещества мозга и прогрессирующую его дегенерацию.
Группа лейкодистрофий включает более 60 форм, которые отличаются видом мутации и возрастом проявления болезни, что влияет на прогноз заболевания. В зависимости от возраста формы могут подразделяться на варианты: инфантильный, ювенильный и взрослый. Все эти заболевания сопровождаются нарушением передачи нервных импульсов в ЦНС, в связи с чем у больных возникают сначала двигательные, а затем и интеллектуальные расстройства, задержка психомоторного развития. По мере разрушения миелина расстройства прогрессируют и за несколько лет у больных развивается тяжелая физическая и психическая деградация. Некоторые формы имеют отличительные симптомы, связанными с поражением и других органов кроме ЦНС. Многие формы настолько редко встречаются, что даже в мировой медицинской литературе описано несколько сотен случаев и наблюдений за больными.
Патогенез
Генетически обусловленный дефект приводит к нарушению обмена (чаще нарушается метаболизм липидов). Это влечет отложение того или иного вещества (метаболита) в тканях. Прежде всего поражается головной мозг и спинной, почки и печень. Токсические метаболиты вызывают разрушение миелина, гибель или атрофию нейронов (клетки нервной ткани), при этом погибшие нейроны замещаются глиальной тканью (соединительная ткань), которая разрастается. Любая лейкодистрофия характеризуется основными патогенетическими процессами: диффузная гибель миелина в полушариях, скопление продуктов, которые образуются после его распада и усиленное разрастание глии. При отдельных формах отмечается специфическая картина — метахроматическое (суданофильное) окрашивание распавшихся продуктов миелина или скопление в зонах распада характерных глобоидных клеток. При болезни Александера в результате мутации в нервной ткани накапливается генетически измененный (мутантный) белок GFAP.
Классификация
Наследственные лейкодистрофии по характеру являются гипомиелинизирующими — миелин изначально не образуется или образуется в недостаточном количестве. Большинство из этих заболеваний наследуется по аутосомно-рецессивному признаку — это значит, что вероятность заболевания у ребенка составляет 25%, если оба родителя-носители гена. В таких случаях с одинаковой частотой в семьях заболевают мальчики и девочки, рожденные от близкородственных браков.
Есть форма, которая характеризуется наследованием, сцепленным с Х хромосомой (адренолейкодистрофия) — она передается Х-хромосомой матери-носительницы болезни. Чаще всего встречаются метахроматическая лейкодистрофия, Пелицеуса-Мерцбахера (или суданофильная), адренолейкодистрофия, болезнь Краббе, Александера и Канавана. Кратко остановимся на этих формах.
Метахроматическая лейкодистрофия — одна из частых и изученных форм. Девочки и мальчики поражаются одинаково часто. При данной патологии отмечается дефицит лизосомного фермента арилсульфатазы. Этот дефект приводит к тому, что в белом веществе головного мозга, периферических нервов и органах (печень, легкие, почки, сердце) накапливаются специфические метаболиты сульфатиды, которые при гистохимическом исследовании дают специфическое метахроматическое окрашивание. Функция внутренних органов не страдает, а в мозговом веществе прогрессируют дегенеративные изменения. Метахроматическая лейкодистрофия по клинике делится на типы: врожденный, поздний инфантильный, ювенильный (ранний и поздний) и взрослый. Все типы протекают с ухудшением двигательной и психической функции, но эти нарушения возникают в разный возрастной период и степень прогрессирования тоже разная.
Врожденная метахроматическая форма развивается до 3-х месяцев и проявляется эпилептическим синдромом и задержкой развития. К ним присоединяется парез и прогрессирующие расстройства глотания. Малыши умирают на первом году жизни.
Поздняя детская форма — самая частая. Заболевание проявляется в 6 месяцев — 4 года. Дети, которые нормально развивались до 8-9 месяцев к году жизни теряют приобретенные навыки — у них нарушается походка, они перестают сидеть и ходить, появляется разболтанность суставов, снижаются сухожильные рефлексы и прогрессирует мышечная слабость, нарушается функция глотания и ухудшается зрение (атрофия зрительных нервов). Постепенно развивается нарушение функции ног, а затем и рук (спастический тетрапарез), нарушения глотания. При хорошем питании через желудочный период продолжается 1-2 года. Но потом двигательная и когнитивная функция быстро прогрессируют, и такие дети умирают в течение 5 лет с начала заболевания.
Ведущий симптом при ювенильной форме (6-10 лет) — нарушение координации движений (мозжечковая атаксия). Больные неустойчивы, ходят неуверенно с широко расставленными ногами, отмечается шаткость при походке. С поздней ювенильной формой больные доживают до взрослого возраста. Взрослая форма проявляется нарушением поведения, из-за чего пациентов часто принимают за психиатрических больных, и снижением когнитивной функции. Постепенно развиваются нарушения движений. Дети редко доживают до десяти лет.
Взрослая форма начинается после пубертата в период до 50-60-лет. Начинается с психических расстройств (психопатия, шизофреноподобный синдром). Также развиваются полинейропатии. Если сравнивать с другими возрастными вариантами, то изменения прогрессируют медленно и заканчивается заболевание тетрапарезом и возрастной деменцией.
Лейкодистрофия Пелицеуса-Мерцбахера связана с генетическим дефектом синтеза апопротеина — белка, который важен для функции клеток олигодендроцитов, участвующих в миелинизации аксонов. Передается по аутосомно-рецессивному признаку: если оба родителя носители мутантного гена в 25% рождается больной ребенок. Второй тип наследования — сцепленная с полом передача (только мальчикам в семье или девочкам).
Проявляется болезнь рано — с 5 до 10 месяцев и имеет медленное развитие. В течение болезни появляется «светлый» промежуток, очень долго длящийся. В связи с этим больные доживают до зрелого возраста и летальный исход возможен только в возрасте 20-30 лет. Болезнь Пелицеуса-Мерцбахера в младенчестве проявляется кивательными движениями головы, блуждающими движениями глаз нистагмом, задержкой развития. Когда малыш начинает ходить, у него появляется атаксия и спастичность конечностей, хореоатетоз (сочетание быстрых и порывистых движений с медленными судорожными). Постепенно развивается нарушение речи и атрофия зрительных нервов. Умственное развитие детей не страдает.
Адренолейкодистрофия связана с мутациями гена АВСD1 и изменением белка ALDP. При генетической диагностике обнаруживают мутации гена ABCD1 (это могут точечные мутации, делеции нуклеотидов и крупные делеции). При этом нарушается транспорт нормальных жирных кислот в миелиновую оболочку и образуются аномальные сверхдлинноцепочечные жирные кислоты в большом количестве, оказывающие токсическое действие на миелин. В результате токсического действия концентрация миелина резко снижается. Помимо поражения ЦНС и периферической нервной системы отмечается поражение надпочечников в виде их недостаточности. В зависимости от преобладания симптомов в клинике выделяют церебральную форму, периферическую и форму только с надпочечниковой недостаточностью.
Церебральная Х-сцепленная адренолейкодистрофия может проявляться в детском возрасте (пик 7-8 лет, детская форма), юношеском (манифестирует в 10-21 лет, юношеская форма) и во взрослом возрасте (30-50 лет, встречается редко). Детская и юношеская формы характеризуются быстрым прогрессированием двигательных нарушений, интеллектуальных и поведения. В детском возрасте наиболее часто встречаются гиперактивное поведение или противоположность ему — аутистическое поведение. У детей возникает агрессивность, снижается память и внимание, возникают проблемы с обучением, нарушается походка и прогрессирует слабоумие.
Значительно реже бывают нарушения зрения и слуха, а также надпочечниковая недостаточность (низкое давление, гиперпигментация кожи, слабость, рвота и тошнота, возникающие периодически). В юношестве прогрессирует умственная отсталость, прогрессивно ухудшается память, развивается спастический тетрапарез (нарушается двигательная функция рук и ног), снижается зрение и слух, появляются судороги. У взрослых заболевание проявляется деменцией и шизофренией. Также у больных нарушается функция глотания и зрения (выпадают поля зрения). Чаще всего прогноз неблагоприятный: несколько лет от начала первых симптомов заболевание прогрессирует и приводит к смерти.
Лейкодистрофия Краббе связана с дефицитом определенного лизосомного фермента, что обусловлено мутациями в гене GALC. При недостатке этого фермента в нервной ткани накапливается галактозилсфингозин — высокотоксичное вещество, вызывающее демиелиенизацию и гибель клеток с образованием в очагах больших глобоидных образований. Болезнь Краббе бывает нескольких подтипов: инфантильный, поздний инфантильный, взрослый и ювенильный. Самая частая из всех форм — инфантильная, она считается классической и составляет до 90% всех случаев.
Первые проявления заболевания наблюдаются в 3-6 месяцев. Сначала у малышей появляется гипервозбудимость, повышенный мышечный тонус, рвота, гастроэзофагеальный рефлюкс, плохая прибавка веса и задержка развития (до этого периода ребенок развивался нормально). На втором этапе заболевания отмечается регресс психомоторного развития (теряются все навыки), появляются судороги и опистотонус, наступает атрофия зрительных нервов (слепота) и прогрессирует гипотрофия (нарушение питания). На третьей стадии заболевания ребенок полностью теряет произвольные движения, и у него возникает децеребрационная поза, которая свидетельствует о тяжелом поражении головного мозга.
 У детей развивается слепота, глухота, он не реагирует на внешние раздражители. Поздняя инфантильная развивается не так быстро, но проявления сходны. Ювенильная формы проявляется в 3-8 лет и характеризуется быстрым психомоторным регрессом всех навыков. Взрослая форма проявляется после 8 лет периферической нейропатией (парестезии, снижение силы мышц, повышение их тонуса). Затем развивается нарушение походки и парезы, отмечается регресс психомоторного развития, потеря зрения. Смертельный исход наступает через 3 года после появления первых симптомов.
У детей развивается слепота, глухота, он не реагирует на внешние раздражители. Поздняя инфантильная развивается не так быстро, но проявления сходны. Ювенильная формы проявляется в 3-8 лет и характеризуется быстрым психомоторным регрессом всех навыков. Взрослая форма проявляется после 8 лет периферической нейропатией (парестезии, снижение силы мышц, повышение их тонуса). Затем развивается нарушение походки и парезы, отмечается регресс психомоторного развития, потеря зрения. Смертельный исход наступает через 3 года после появления первых симптомов.
Лейкодистрофия Канавана обусловлена мутацией гена ASPA, ответственным за синтез фермента аспартоацилазы. Этот фермент расщепляет токсичный N-ацетиласпартат. Накопление токсичного вещества вызывает дегенерацию серого и белого вещества мозга. Тип наследования аутосомно-рецессивный с передачей гена от обоих родителей.
Болезнь Кэнэвэн-ван-Богарта-Бертрана развивается постепенно и сначала ребенок развивается нормально. Через несколько месяцев навыки начинают угнетаться, снижается мышечная активность, увеличивается спастичность мышц, взгляд становится нефиксированным, затрудняется вскармливание из-за нарушений глотания, а также постепенно увеличивается объем головы. Болезнь прогрессирует стремительно, но произвольные движения пока сохраняются. Потом ребенку становится трудно фиксировать голову, развиваются спастические парезы рук и ног. По мере прогрессирования глотательный рефлекс утрачивается и ребенка кормят через пищеводный зонд. Развиваются судорожные припадки, реакции на раздражители отсутствуют и утрачивается зрение — эти симптомы являются пиком заболевания. Развитие симптомов может быть быстрым (в течение нескольких месяцев) и длительным (до 10 лет). Бактериальные или вирусные инфекции — частые осложнения этого заболевания.
Причины
Как было указано выше, причиной этой группы заболеваний являются генные мутации и наследование патологии от родителей. Вид мутаций установлен для наиболее распространенных форм. Мутации выявляются в генах, кодирующих различные лизосомальные ферменты:
- при адренолейкодистрофии выявляются мутации гена АВСD1;
- болезнь Краббе — в гене GALC длинного плеча 14 хромосомы;
- метахроматическая лейкодистрофия — в гене 22 хромосомы, отвечающего за синтез фермента арилсульфатазы А;
- болезнь Канавана–ван-Богарта–Бертранда — аномальный ген ASPA;
- болезнь Пелициуса–Мерцбахера — мутации ген PLP1.
Симптомы
В зависимости от формы лейкодистрофия у ребенка проявляется или в грудном возрасте (первые месяцы) или раннем детстве. До этого времени ребенок развивается нормально и не отстает от сверстников. Постепенно развивающиеся изменения в ткани мозга (головного или спинного) проявляются неврологическими расстройствами.
Первыми появляются двигательные нарушения: ухудшается координация, ребенку трудно удерживать тело в равновесии, он перестает сидеть и ходить. Развивается мышечная слабость, изменяется тонус мышц (чаще всего он повышается, но может снижаться), появляются мышечные подергивания, а потом судорожные приступы. К двигательным расстройствам присоединяются психические нарушения (меняется поведение), ухудшается интеллект и память. Вышеперечисленные симптомы неуклонно прогрессируют. Постепенно ухудшаются слух и зрение. На поздних стадиях развиваются параличи, выраженная олигофрения, слепота, глухота, ребенок теряет возможность глотать. В терминальной стадии наблюдается децеребрационная ригидность. Больной лежит в позе разгибания. При этом шея и позвоночник напряжены, а голова запрокинута, ноги разогнуты и повернуты внутрь, могут быть скрещены. Стопы и руки разогнуты, а пальцы сжаты в кулак. Скорость прогрессирования симптомов заболевания больше, чем раньше проявились первые симптомы. Периферическая нейропатия у ребенка — важная черта метахроматической лейкодистрофии и болезни Краббе, при которых нарушается метаболизм липидов миелина.
Анализы и диагностика
- Анализируется семейный анамнез и выясняется наличие данного заболевания у близких родственников.
Генетическое тестирование — ключевое в диагностике этой группы заболеваний. Молекулярно-генетическое обследование больного ребенка предусматривает анализ генов, входящих в панель «Нейродегенеративные заболевания». - Определение уровней ферментов, синтез или транспорт которых нарушен при том или ином заболевании. Биохимические маркеры заболеваний отличаются от нормы в несколько десятков раз. При болезни Краббе снижена активность фермента галактоцереброзидазы в лейкоцитах крови. При адренолейкодистрофии повышается в крови уровень жирных кислот с очень длинными цепями (сокращенно VLCFA). Жирные кислоты повышены уже на момент рождения ребенка. При метахроматическом варианте определяют сульфатиды в крови (уровень повышен) и активность арилсульфатазы А в лейкоцитах (отмечается сниженный уровень этого фермента).
- При электронейромиографии — снижение проведения импульса по нервам на периферии (Краббе и метахроматическая форма).
- При многих формах в спинномозговой жидкости определяется высокий уровень белка за счет разрушения клеток мозга.
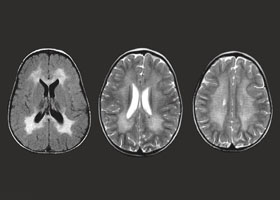
- При проведении МРТ в разной степени отмечаются очаговые изменения белого вещества разных отделов мозга (подкорковых структур, мозжечка, мозолистого тела, пирамидных трактов и так далее).
- При метафроматической форме на начальных стадиях гиперинтенсивные очаги локализуются в перивентрикулярных областях. На поздней стадии обнаруживается атрофия мозолистого тела и больших полушарий.

Доктора



Лечение
До настоящего времени не разработано лечение лейкодистрофий, которое бы сдерживало прогрессирование. Однако, симптоматическое лечение в какой-то степени уменьшает клинические проявления. Сюда включается противосудорожная и дегидратационная терапия, массаж и ЛФК для улучшения состояния мышц и функции движения. Значение придается также эрготерапии — сохранение и поддержание бытовых навыков и самообслуживания. Предпринимаются попытки консервативного лечения при адренолейкодистрофии применением глицеротриолеатного масла, которое снижает уровень длинноцепочечных жирных кислот на 30-40%.
Также назначается «масло Лоренцо» — смесь олеиновой и эруковой кислот (соотношение 4:1). Прием этого масла внутрь в течение месяца нормализует уровень жирных кислот в крови. Показан прием его с профилактической целью на доклинической стадии до развития неврологических симптомов у бессимптомных пациентов. Также есть смысл принимать его для замедления прогрессирования. Действительно, положительный эффект получают при применении на доклинической стадии, но оно неэффективно у больных с выраженными нарушениями. Также неэффективным оказалось применение гиполипидемических средств (Ловастатин, Симвастатин), поскольку снижение уровня длинноцепочечных жирных кислот незначительно.
Больным с адренолейкодистрофией назначается низкожировая диета. Больным с надпочечниковой недостаточностью под контролем гормонов назначается заместительная стероидная терапия. Предпринимаются попытки поиска препаратов, которые могли бы активировать аномальные гены, но они пока безуспешны. Методы генной терапии основаны на клонировании и внедрении здорового гена, который заменяет патологический ген. Лечение носит экспериментальный характер — непатогенные вирусы-носители искусственно синтезированного гена водятся вместе с раствором в головной мозг.
Процедуры и операции
Методом лечения данной неврологической патологии является донорская трансплантация костного мозга. Если она успешно проведена, достигается нормализация уровня недостающих ферментов и обмена веществ, соответственно улучшается качество и продолжительность жизни больных. Основным условием является проведение трансплантации до развития неврологических нарушений. Трансплантация не исправляет уже развившиеся поражения ЦНС — только замедляет или останавливает их прогрессирование.
При быстро прогрессирующих формах трансплантацией не удается улучшить состояние и избежать тяжелой инвалидизации больного. Это объясняется тем, что после трансплантации в течение 1-2 лет донорские клетки еще «не работают» в полную силу и могут полностью нормализовать функцию миелина. При медленном развитии болезни трансплантация более успешна. После трансплантации возможны осложнения — отторжение трансплантата, инфекционные заболевания и реакция «трансплантат против хозяина». Если трансплантацию нет возможности выполнить лечение направлено на облегчение симптомов.
При невозможности питания организуется кормление через гастростому или назогастральный зонд. В случае выраженной дыхательной недостаточности больной организуется респираторная поддержка — наложение трахеостомы и искусственная вентиляция легких.
Диета
Из всех видов лейкодисторофий диетотерапия назначается при адренолейкодистрофии в сочетании с применением масла Лоренцо. Больным рекомендуется ежедневное ограничение поступления жиров на 15%. Исключаются жирное мясо, салями, сливочное масло, маргарин, растительное масло, рыбные консервы в масле, рыбий жир, жирная рыба, копченая рыба, молоко высокой жирности, орехи, семена, оливки, авокадо, мороженое, жирные пудинги и десерты, шоколадная паста, арахисовое масло, какао, шоколад.
Разрешается постное мясо (говядина, ягненок, индейка, курица), белая нежирная рыба, фрукты, фасоль, чечевица, горох, рис, кукуруза, хлеб, макароны, мучные изделия, шербет, меренга, сахар, джем, мед, мармелад, чай, сок, кофе. Параллельно с этим ребенок должен ежедневно принимать масло Лоренцо от 30 мл до 55 мл в зависимости от возраста.
Профилактика
- Исследование генетической панели родителей дает возможность определить генные мутации.
- Выявление женщин-носительниц адренолейкодистрофии проводится биохимическим определением уровня длинноцепочечных жирных кислот VLCFA в крови. Будущие родители из группы риска по метахроматической форме заболевания должны обследовать активность арилсульфатазы А.
- В семьях, где были случаи рождения детей с этой патологией, проводится пренатальная диагностика при всех последующих беременностях. Данный вид диагностики разработан для метахроматической лейкодистрофии, адренолейкодистрофии и глобоидно-клеточной дистрофии. При метахроматической лейкодистрофии определяют арилсульфатазу А в клетках амниотической жидкости или ворсинах хориона. Пренатальная диагностика адренолейкодистрофии проводится анализом ДНК.
Последствия и осложнения
- Вторичная гидроцефалия.
- Рецидивирующие инфекции и тяжелые пневмонии на фоне обездвиженности.
- Тяжелые двигательные нарушения вплоть до развития децеребрационной ригидности.
- Интеллектуальные расстройства до глубокого слабоумия.
- Нарушения глотания.
- Слепота.
- Глухота.
- Вегетативное состояние.
- Смерть больного.
Прогноз
Прогноз при лейкодистрофии неблагоприятный. Особенно тяжело и быстро протекают церебральные формы заболевания с ранним появлением в детстве. Некоторые перспективы дает проведение трансплантации костного мозга, которое останавливает/замедляет ход болезни, а также позволяет сохранить двигательные функции и интеллект. Однако, важнейшее условие — своевременное ее проведение еще до начала развития неврологических проявлений.
Список источников
- Новиков П. В., Михайлова С. В., Захарова Е. Ю., Воинова В. Ю. Федеральные клинические рекомендации по диагностике и лечению Х-сцепленной адренолейкодистрофии. 2013.- 19 с.
- Михайлова С. В., Захарова Е. Ю., Петрухин А. С. Нейрометаболические заболевания у детей и подростков: диагностика и подходы к лечению (2-е издание переработанное и дополненное). М.: Литтерра, 2017; 313-337
- Айкарди Ж. Заболевания нервной системы у детей. М.: Издательство Панфилова, издательство Бином, 2013; 1: 372.
- Царева Ю. А., Зрячкин Н. И., Кузнецова М. А., Рядченко А. В. Лейкодистрофия Краббе (Обзор литературы с описанием клинического случая)/ Российский педиатрический журнал. 2018. - Т. 21. № 2. С. 114-120.
- Фазлеева Л. К., Поладова Л. В., Шагиахметова Д. С., Аюпова В. Г. Болезнь Краббе - трудности диагностики и терапии / Практическая медицина. 2010. - № 7 (46). С. 128.
Комментарии






Статьи по теме





Последние комментарии
Инна: Что-то меня дернуло и решила я попробовать новый гель для интимной гигиены. Баночка такая ...
Вероника: Все понятно,но действительно что при ПН можно например рис,компот,мед при диабете ...
Елизавета: Несколько месяцев назад, спустя почти 2 года свободы, у меня появились отношения. Вместе ...
Людмила: Гусиная кожа это не заболевание, а состояние кожи, и улучшить его можно увлажнением ...